Abstract Troubleshooting HPLC Instrumentation and separations requires a fundamental understanding of how the instrument functions and how the separation works. This chapter provides a practical guide to common HPLC problems, along with more in-depth information to help the reader understand the relationships between the observed symptoms and the underlying causes. The practical approach presented here is meant to serve as both a troubleshooting guide and an HPLC learning tool.
KeywordsThis article was written by Lee Polite and Harold McNair.
LevelBasic
This chapter provides an overview of HPLC troubleshooting and summarizes guidelines for system maintenance. Troubleshooting problems are classified into four major categories:
- Problems observed on the chromatogram
- Operating parameters
- Leaks
- Pressure problems.
After each problem is described, symptoms and possible solutions are ![]() proposed.
proposed.
Problems Observed in ![]() Chromatograms
Chromatograms
|
| Symptom | Probable Causes |
| Chromatography. |
No peaks.
| No flow, poor injection, system down, bad data cables |
| Need more help? Link to 'Peak problems' . | Too many peaks | Carryover (auto sampler) |
|
| Fronting peaks | Column over loaded |
|
| Tailing peaks | Dirty frit or column, |
|
| Change tR | Flow rate, mobile phase composition |
| Need more help? Link to 'Baseline problems' | Noisy baseline | Pump problems, dirty column, old lamp |
| Leaks | Column leaks | Loose, dirty, or wrong fittings |
|
| Pump Leaks | Loose fittings, pump, |
|
| Injection Leaks | Rotor seal failures |
Pressure | High Pressure | Blockage of frit or column or connections |
| Need more help? Link to 'Pressure problems' | Too Low Pressure | Leaks, low flow rate, channel in column, pump problemns |
|
| Pressure Cycling | Check valves, pump seal, air bubbles |
Poor Resolution | N value Low | Old column, dirty column, dirty matrix sample size too large, bad fittings or tubing |
| Need more help? Link to resolution problems | a value Low | Wrong stationary phase |
|
| k value too low | Too strong mobile phase |
| Bad column stability | Need more help? Use this link. | |
With current data systems a variety of chromatographic performance characteristics can easily be recorded and printed out. These include retention times, ![]() peak areas, peak heights, asymmetry factor, plate counts, selectivity values and resolution. Major changes in any of these parameters may (probably will) require action on your part.
peak areas, peak heights, asymmetry factor, plate counts, selectivity values and resolution. Major changes in any of these parameters may (probably will) require action on your part.
This is a basic introduction to peak problems. Click to go to detailed tables on peak problems
No Peaks
No peaks usually indicate an instrumental problem. No peaks "No peaks" could be due to a number of causes including no sample injected, system not turned on properly, major leaks, a dead detector, wrong mobile phase, or a particularly retentive or adsorptive column.
"No peaks" could be due to a number of causes including no sample injected, system not turned on properly, major leaks, a dead detector, wrong mobile phase, or a particularly retentive or adsorptive column.
- Check and measure (use a stop watch and graduated cylinder) flow rate. No flow usually indicates pump problems, flow system blockages or empty solvent reservoirs.
- Then check for leaks; if any eliminate
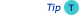 them.
them.
Too Many Peaks
usually this occurs from sample carryover, usually in the auto sampler or on the column. Using a sample solvent stronger than the mobile phase, may cause sample precipitation in the column. The next blank run will show the original chromatogram, but usually at a lower concentration.
Too Many ( extra) peaks Clean the column!
Clean the column! ![]() Extra peaks could also result from the degradation of an unstable component. Investigate the stability of suspected peaks with respect to time, pH and light. After the first injection, some sample vial septa may produce an extra peak in subsequent injections from the same vial. This is usually a plasticizer extracted from the septa. Change the type of septa.
Extra peaks could also result from the degradation of an unstable component. Investigate the stability of suspected peaks with respect to time, pH and light. After the first injection, some sample vial septa may produce an extra peak in subsequent injections from the same vial. This is usually a plasticizer extracted from the septa. Change the type of septa.
Gradient Elution.
Run a ![]() blank gradient – if the extra peaks still occur they are probably impurities in the solvent. Make up new mobile
blank gradient – if the extra peaks still occur they are probably impurities in the solvent. Make up new mobile ![]() phase.
phase.
Too few peaks
This is rare and most often indicates poor resolution, i.e. one or more peaks are overlapping (unresolved) from other ![]() peaks.
peaks.
Loss of some peaks or changes in peak area may also indicate a "bad" or active column which irreversibly adsorbs the peak, often a basic peak with silica based columns.
Fronting and Tailing Peaks
Different peak shapes 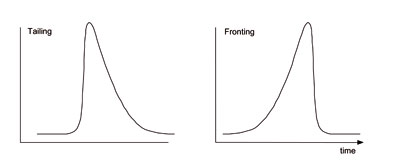 Fronting peaks
Fronting peaks
are usually caused by column overload (sample amount exceeding the sample capacity of the column); dilute the sample 1/10 and re-inject.
Tailing peaks
are caused by secondary (undesirable) interactions of the analyte with the stationary phase, or a poor connection (excess void ![]() volume).
volume).
Check to make sure all fittings and tubing between the injector and detector are correct.
Asymmetric peaks shapes can also be caused by degradation of the analyte during chromatography.
Negative and Positive Peaks
Negative and positive peaks or dips in the chromatogram are common with an R.I. detector. Negative peaks at the solvent front are caused by refractive index changes with UV detectors or by sample solvents with less absorbance than the mobile phase. These baseline perturbations are usually ignored by starting data integration after the solvent front. If all peaks are negative, it might be an indication of the wrong polarity in the detector output ![]() signal.
signal.
Broad Peaks and Split Peaks
Broad peaks and split peaks are indications of degraded column performance caused by sample contamination, a partially blocked inlet frit, or column voids caused by dissolution of silica particles, usually at high pH. Replace the ![]() column.
column.
Anomalous peak shapes can also be caused by injecting samples dissolved in solvents stronger than the mobile phase. If possible, the sample solvent should be of weaker strength than the mobile phase or of equal strength to the mobile phase. If strong solvents must be used, the volume should be kept small (e.g., < 5 mL) to prevent peak broadening or peak splitting of the early eluting peaks.
Click to go to detailed tables on peak problems
Changes in Retention Time 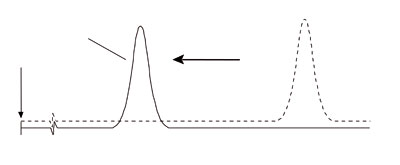 Changes in flow rate
Changes in flow rate ![]() and mobile phase composition are the major factors responsible for changes in retention
and mobile phase composition are the major factors responsible for changes in retention ![]() time.
time.
Organic solvents change the pH. Table 1 lists the most common buffers used to adjust pH.
| pH value | Buffer/modifier | UV cut-off (nm |
| 2.0-3.0 | Phosphate | 210 |
| 2.5-7.5 | Citrate | 250 |
| 3.5-6.0 | Acetate | 230-240 |
| 6.0-8.5 | Phosphate | 210 |
| 7.0-9.5 | "TRIS" (tris-hydroxymethylaminomethane) | 220-225 |
| 8.0-10.5 | Borate | 210 |
| 9.0-12.0 | Diethylamine (fresh!) |
|
Wrong pH
The buffer could be too weak; use a 20-100 mM buffer when possible (note: you may want to limit this to 50mM). Never chromatograph weak acids or weak bases with a pH close to their pKa (they will be only partially ionized and may show tailing or badly shaped peaks in addition to irreproducible retention times). Use a pH + 2 units above or below analyte pKa for good reproducibility.
Wrong mobile phase
Too much organic (methanol or acetonitrile) in R.P. separations will cause a decrease in all retention times. Make new mobile phase.
In gradient elution, lack of equilibration between runs often produces shorter retention times only for early peaks. Confirm equilibrium by measuring retention time stability by making 3 or 4 replicate runs. High organic or aqueous content (>95%) causes some phase collapse and may require longer equilibration times between gradient runs. Wait longer between runs or start with a stronger mobile phase initially.
Wrong flow rate. There is an optimal flow rate for each column, typically about 1.0 ml/min for a 4.6 mm I.D. column. Retention time is inversely proportional to flow rate, so measure, and adjust if necessary, the flow rate. An easy way to shorten the analysis times for most methods is to increase the flow rate to 1.5 or 2.0 mL/min if the pressure allows.
Decreasing retention time with fronting peaks often indicates an overloaded column. Dilute the sample 1/10 and re-inject. Overloaded column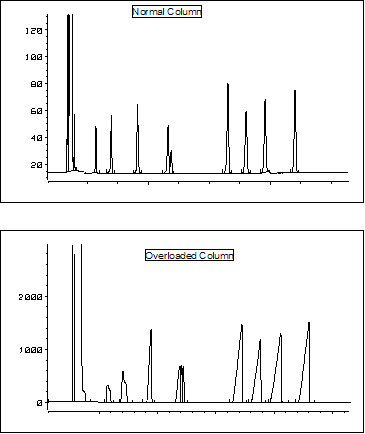
Retention time stability only after a few initial injections indicates active sites in the column. One possibility is that a polar molecule in the sample temporarily covers (adsorbs onto) these sites. Purchase a more inert column, or add a mobile phase modifier like triethylamine to cover up active silanol sites.
Click here to go to more details on retention time problems
Reproducible peak areas are essential for good LC performance. Expected precision is a function of analyte activity, sample concentration, detector, flow rate stability and data system. Usually one can expect a % RSD's of 1% or less for 6 replicate injections of a standard of reasonable concentration down to about 10 ppm (wt./vol.). Lower concentrations often exhibit higher RSD's due to problems in precision of injection techniques and/or LOD's of the detector and data system.
| Peak retention time precision | ||
| Þ with oven | < 0.3% | |
| Þ without oven | < 0.7% | |
| Peak area precision | <1.5% |
Peak area changes may also be due to changes in volume injected, flow rate, wavelength of detector, pH, leaks, sample stability, integration problems, and partial loss of sample due to irreversible adsorption on a dirty frit or active column.
Time spent in method development in choosing a proper column, pH, mobile phase, even column temperature to produce symmetrical peaks usually results in better precision of peak areas.
If only one peak in a mixture shows poor precision, possible causes could be: wrong pH, (analyte is only partially ionized); large peak asymmetry causing integration errors (try a more inert column, or a mobile phase modifier). If retention factor ( k), is too low (<2) the peak may be overlapping with other peaks. Decrease the strength of mobile phase.
Click here for more peak problems
If all peaks are broad, the column efficiency (N) may be too low. Try a longer column or smaller particle diameter or use the optimal flow rate.
Table 3 shows plate number N as a function of column length and particle diameter.
| Length (CM) | Particle Diameter um | Efficiency N |
| 25 | 10 | 10,000 |
| 25 | 5 | 22,000 |
| 15 | 5 | 14,000 |
| 10 | 5 | 10,000 |
| 10 | 3 | 12,000 |
*Experimental data – H. McNair's Lab
Click here for more examples on asymmetrical peaks
Short Term Noise, if detector related is usually very small. Short term (detector) noise
Modern UV detectors have noise specifications of about + 1 x 10-5 AU. More noisy signals often originate with the low energy of an aging UV lamp. UV lamps should be replaced regularly, after a specified number of operating hours, or after a certain time period. Some short-term noise is normal at high sensitivity.
Synchronous Noise is a periodic noise usually associated with pump strokes.
Synchronous noise With a piston volume of 100 ml, a flow rate of 1.0 ml/min will generate noise spikes every 6 seconds. In order to verify that the noise is caused by the pump, turn off the pump. If these periodic noises disappear, they came from the pump. Pump malfunctions, leaking check valves, leaking pump seals, a faulty pulse dampener, or air bubbles are also possible causes. Air bubbles passing through the pump is the most frequent cause of synchronous or periodic noise. Modern LC systems use in-line vacuum degassing. This is the best way to avoid air bubbles. Helium sparging is not as effective and requires a helium cylinder. Helium does prevent bacterial growth in buffers.
With a piston volume of 100 ml, a flow rate of 1.0 ml/min will generate noise spikes every 6 seconds. In order to verify that the noise is caused by the pump, turn off the pump. If these periodic noises disappear, they came from the pump. Pump malfunctions, leaking check valves, leaking pump seals, a faulty pulse dampener, or air bubbles are also possible causes. Air bubbles passing through the pump is the most frequent cause of synchronous or periodic noise. Modern LC systems use in-line vacuum degassing. This is the best way to avoid air bubbles. Helium sparging is not as effective and requires a helium cylinder. Helium does prevent bacterial growth in buffers.
Asynchronous noise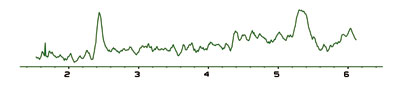 Asynchronous Noise is random noise often associated with contamination of mobile phases, a dirty or leaky detector cell, poor mixing of gradients, sample degradation, leaks or late eluting impurities in samples or solvents. Once again, turn off the pump to verify that it is not a pump problem.
Asynchronous Noise is random noise often associated with contamination of mobile phases, a dirty or leaky detector cell, poor mixing of gradients, sample degradation, leaks or late eluting impurities in samples or solvents. Once again, turn off the pump to verify that it is not a pump problem.
Very fast synchronous or non-synchronous noise spikes are caused by electrical problems such as power/voltage fluctuations, poor grounding or loose detector/ data system connections. Electronic spikes
If not obvious, the best remedy is to call a service engineer (see the last page for routine items to check before calling the service engineer). These spikes could also be due to air bubbles in the detector cell, less likely with the new generation of in-line vacuum degassing systems.
- Baseline Drift: Baseline drift often reflects lack of temperature equilibrium, or changes in the energy output of the UV lamp. This baseline drift is reduced significantly in dual-beam or reference wavelength absorbance systems. Baseline shifts associated with gradients are normal, indicating more absorption in the stronger solvent (particularly when methanol is used as the strong solvent). Non-specific drifts, up and down, can be caused by strongly retained peaks slowly eluting off a contaminated column.
Click here for more examples of problems with noise.
With the exception of normal phase and GPC, all other forms of HPLC include water in the mobile phase. In almost all cases the pH should be carefully controlled. If no buffer is used, the pH of the water component can easily drift due to absorption of CO2 from the atmosphere or due to extraction of impurities from sample loops, column frits, and dirty columns.
Ammonium phosphate is a very popular buffer for several reasons:
- It's pH range of 2.0 – 3.0 neutralizes all weak acids, and fully ionizes all weak bases; thus it is versatile.
- It is readily available in high purity
- It has a high solubility with high concentrations of acetonitrile; i.e. no precipitation
- Its UV cut-off is about 210 mm. Phosphate buffers should not be used however, when interfacing to a LC/MS system because they will contaminate the ion source. For LC/MS, use the more volatile acetate buffers.
As mentioned earlier, chromatography with pH close to the pKa of any analyte is to be avoided. Use a pH + 2 units removed from the pKa.
Dissociation curves for weak acids and bases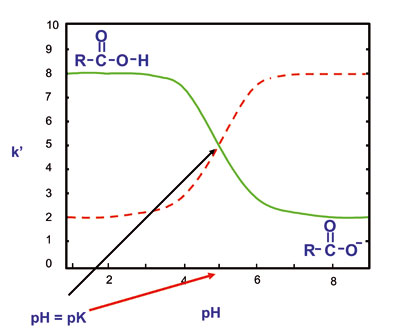
Addition of organic mobile phase changes the pH of aqueous solvents. It is recommended to measure pH before adding organic solvents. Remember buffers almost always increase UV absorption, which could be a problem in gradient elution. Buffers are an enemy of pump seals (if precipitation occurs), and buffers shorten the life-time of silica based columns.
In theory, pH affects only weakly ionized acids and bases; neutral molecules and strong acids and bases are not affected. However, pH can affect the stationary phase if it is silica based. To minimize the effect of high pH on silica gel based columns: (1) work at room temperature; (2) use a densely bonded, highly end capped reverse phase column; and (3) use a high percentage of organic component in the mobile phase as this lowers the "effective" pH.
Silanols –on silica surface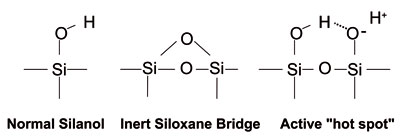 The silica surface has silanol groups which are polar and weakly acidic. In normal phase, (pure silica) these silanols are responsible for the necessary polar interactions resulting in separations. In bonded phase packings, both normal and reversed phase, columns still contain unreacted silanols groups. Less than 50% of all silanols can be reacted with organo chlorosilanes to form bonded phases. Unless thoroughly end-capped or one of the newer "hybrid" columns are used, these silanols often cause problems with basic compounds. To check for silanol activity, inject pyridine and examine the peak shape. Only a well deactivated column will produce a symmetrical peak. It may be necessary to use a low pH, 2-3, in order to neutralize any free silanol groups.
The silica surface has silanol groups which are polar and weakly acidic. In normal phase, (pure silica) these silanols are responsible for the necessary polar interactions resulting in separations. In bonded phase packings, both normal and reversed phase, columns still contain unreacted silanols groups. Less than 50% of all silanols can be reacted with organo chlorosilanes to form bonded phases. Unless thoroughly end-capped or one of the newer "hybrid" columns are used, these silanols often cause problems with basic compounds. To check for silanol activity, inject pyridine and examine the peak shape. Only a well deactivated column will produce a symmetrical peak. It may be necessary to use a low pH, 2-3, in order to neutralize any free silanol groups.
Use only HPLC grade solvents. This is the only standard acceptable for the exacting requirements of modern HPLC systems. Record the lot number in case a problem of quality does arise.
If you use an in-house HPLC grade water system, compare the quality of that water with a commercial grade of HPLC water. Pump 100 ml of your in-house water through an R.P.-18 column (this is your sample); all of the organic impurities are adsorbed on the column. Now program the methanol concentration from 0-90% in 9 minutes and record the peak area with a 254 mm UV detector. There will be a large ugly peak due to the large amount of organic contaminants in 100 ml of water. Compare this peak area to that of the commercial grade water. Use the water which has the fewest impurities.
Particle Diameter – (dp)
The particle diameter affects many chromatographic problems but is not a source of problems in itself. The quality of current day LC packings is outstanding. Smaller particles provide more plates (N) and result in better resolution (refer to Table 2). However, the pump pressure required to maintain flow rate is inversely proportional to the particle diameter squared, thus you will rarely see a long column packed (>15 cm) with small particles (< 3.5 um) unless you have a high pressure pump. Particle size 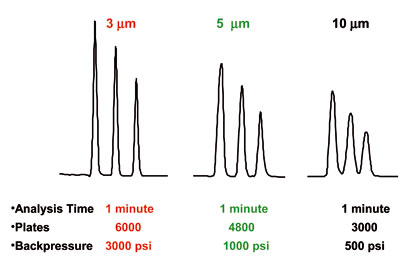
Column Length
Both plate number N and pressure drop are proportional to column length. Twenty years ago most columns were 25 cm in length packed with 10 mm particles. As high quality smaller particles (no easy task to manufacture) became available, column lengths became shorter. Since in isocratic mode analysis time is proportional to column length, the current trend is for faster analyses using shorter columns packed with smaller particlesColumn Length 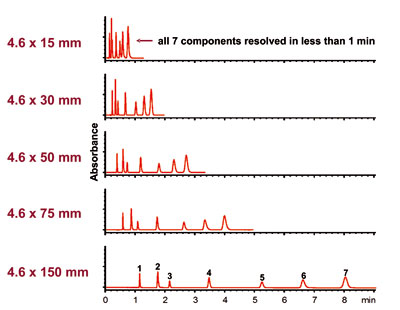 .
.
Flow rate
Van Deemter type plots (discussed earlier in Chapter 2) show the effect of linear velocity on band broadening (HETP). Each column has an optimal flow rate to produce a minimum in band broadening. The figure hereunder shows a typical such plot for 10, 5 and 3 mm particles. Note that this is a fairly minor effect with small particles (1.8, 3.0, 3.5, and even 5 micron particles). Van Deemter plot 10,5 and 3 u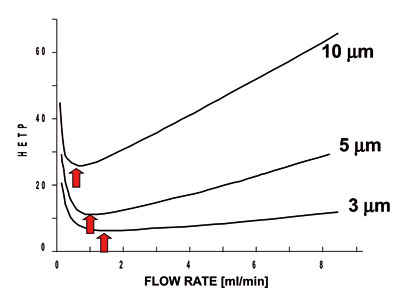
Optimal flow rates are approximately 0.8, 1.2 and 2.5 mL/min for the 10, 5 and 3 mm columns. Thus, smaller particles show faster optimal flow rates. The interesting part however is the slope of each plot after the minimum. Smaller particles allow faster than optimal flows with minimal loss in efficiency.
Column leaks are a routine, almost normal occurrence in HPLC; fortunately, they are easily resolved. Since columns, are being replaced on a regular basis, column leaks, usually at the inlet side (higher pressures) are the most common problem. We recommend using PEEK ferrules and universal (finger tight) nuts.
HPLC Fittings
First make sure the column fittings are tight. If they are tight and leaks still occur, loosen the fitting and start again. If you still have leaks, replace that fitting and the associated hardware. Replace stripped or dirty fittings with new fittings.
No visible leak, but white powder (residue of buffers) at any system connection indicates a leak. First, tighten; then if necessary cut the tubing and replace all hardware.
Pump leaks arise from loose check valves (tighten, if necessary replace), loose inlet or outlet fittings, pump seal failure, pressure transducer failure, or purge valve failure. With a proper manual and minimal training most workers can replace check valves, filter, pump seals, detector lamps, flow cells, auto sampler syringes and manual injector rotor seals. Most pharmaceutical laboratories will have service contracts that provide both emergency and routine maintenance service.
Injector leaks usually occur because of loose connections, dirty or plugged sample loops, rotor seal failure, an improper or broken syringe, wrong syringe needle type and/or blocked or elevated waste line.
Detector leaks arise from cell gasket failure, loose connections, blocked outlet line, or a blocked or dirty flowcell.
Pressure too high is most often caused by partial blockages (listed in order) in filters, column inlet frits, sample loop, column outlet frit or detector. To isolate the problem, disconnect all components coming after the pump in the flow system. Add one at a time, in the proper order: connecting tubing, sample valve (or auto sampler), guard column, analytical column, detector, any post detector device (second detector, split valve or sample collection device). Repair or replace that component responsible for the high pressure.
Columns with small particles (3 mm and smaller) generate higher than normal pumping pressures; that is why they are usually short in length, 5 or 10 cm.
Smaller particle columns also develop blockages earlier than larger particles, so smaller inline filter (0.5 mm I.D) are commonly used. If the high pressure blockage occurs in the column, the most likely culprit is the column inlet frit. Clean or replace this frit if possible. If the column itself is the problem, disconnect the column from the detector, reverse the direction of flow (most blockages are in the column inlet side) and flush with 50/50 water and methanol at 60 C. This elevated temperature is used to more easily dissolve precipitates of buffers and samples. After 10 to 20 column volumes increase to stronger solvents, i.e. 90% methanol, then 90% Tetrahydrofuran (THF).
Low, or no pressure is caused by leaks (piston seal, column connections, injector), pump malfunctions (lost prime, air bubbles in pump head, vapor lock, faulty check valves, broken piston), or inadequate solvent supply (empty solvent reservoir, plugged solvent sinker, crimped solvent lines or wrong solvent mixture).
Pressure cycling is caused by: air bubbles in check valves (remedied by degassing the solvent; malfunctioning of check valves or pulse dampeners; or partial system blockages.
Click here for more examples on pressure problems.





