LevelBasic
The most widely used technique of sampling with solid phase microextraction consists of exposing a small amount of extracting phase (coating) associated with a fibre to the sample, for a predetermined amount of time
1. Sample preparation with SPME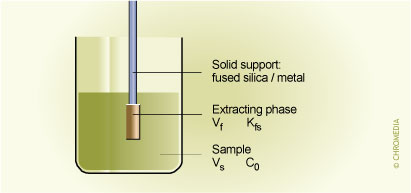
Fig.1. Vf = volume of fibre coating; Kfs = fibre/sample distribution coefficient; Vs = volume of sample; C0 = initial concentration of analyte in the sample.
Typically, the microextraction process is considered complete when the analyte concentration has reached distribution equilibrium between the sample matrix and the fibre coating. The equilibrium conditions can be described by equation (1), according to the law of mass conservation, if only two phases are considered (for example, the sample matrix and the fibre coating):3
![]() (1)
(1)
The distribution coefficient Kfs of the analyte between the fibre coating and sample matrix is defined as:
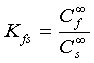 (2)
(2)
Equations (1) and (2) can be combined and rearranged into:
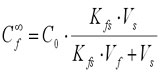 (3)
(3)
Finally, the number of moles of analyte n extracted by the coating can be calculated from equation (4):
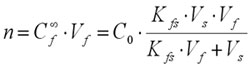 (4)
(4)
Equation (4) indicates that the amount of analyte extracted onto the coating (n) is linearly proportional to the analyte concentration in the sample (C0), which is the analytical basis for quantification using SPME.
Equation (4), which assumes that the sample matrix can be represented as a single homogeneous phase and that no headspace is present in the system, can be modified to account for the existence of other compartments in the matrix, by considering the volumes of the individual phases and the appropriate distribution constants. In addition, when the sample volume is very large, i.e. Vs >> Kfs·Vf , equation (4) can be simplified to:
![]() (5)
(5)
which points to the usefulness of the technique when the volume of the sample is unknown. In practice, the fibre can be exposed directly to the flowing blood, ambient air, water, etc. The amount of extracted analyte will correspond directly to its concentration in the matrix, without depending on the sample volume.
The amount of analyte extracted onto the fibre coating is at a maximum when the equilibrium is reached, thus achieving highest sensitivity. If sensitivity is not a major concern of analysis, shortening the extraction time is desirable. In addition, the equilibrium extraction approach is not practical for solid porous coatings, due to the displacement effect at high concentrations. For these circumstances, the extraction is stopped and the fibre is analyzed before the equilibrium is reached. The kinetics of absorption of analytes onto a liquid fibre coating can be described as:4
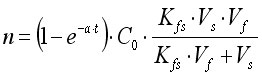 (6)
(6)
where t is the extraction time, and a is a time constant, representing how fast an equilibrium can be reached. When the extraction time is long, equation (6) becomes equation (4), characterizing equilibrium extraction. If the extraction equilibrium is not reached, equation (6) indicates that there is still a linear relationship between the amount (n) of analyte extracted onto the fibre and the analyte concentration (C0) in the sample matrix, provided that the agitation, the extraction time, and the extraction temperature remain constant.
As equation (5) indicates, the extraction process is dependent on the distribution constant Kfs. This is a characteristic parameter that describes the properties of a coating and its selectivity toward the analyte versus other matrix components.
Because of its solvent-free nature and the small size of the fibre coating, SPME can be interfaced conveniently to analytical instruments of various types. Only extracted analytes are introduced into the instrument, since the extracting phase is non-volatile and insoluble in most organic solvents. Thus, there is no need for complex injectors designed to deal with large amounts of solvents, and these components can be simplified for use with SPME.1,5 Depending on the method of subsequent analysis, the sensitivity of determinations using the SPME technique is very high, facilitating trace analysis.6 Although in most cases the analytes are only partly extracted from the sample, all extracted material is transferred to the analytical instrument, resulting in good performance. Carryover should be checked for each analyte and the desorption conditions should be chosen so that the analyte remaining on the fibre is less than 0.1% of the initial amount. The solvent free process results in narrow bands reaching the instrument, giving taller, narrower peaks and better quantification.
Fig.2 describes an SPME device based on the HamiltonTM 7000 series microsyringe:2. Custom-made SPME based on Hamilton 7000 syringe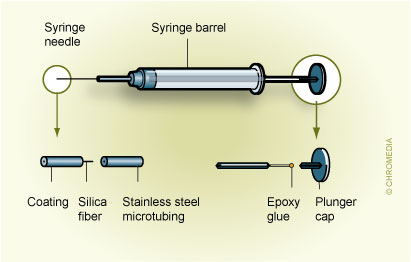 Movement of the plunger allows exposure of the fibre during extraction and desorption and its protection in the needle during storage and penetration of the septum. A commercial SPME device is illustrated in Fig. 3.
Movement of the plunger allows exposure of the fibre during extraction and desorption and its protection in the needle during storage and penetration of the septum. A commercial SPME device is illustrated in Fig. 3. 3. First commercial SPME device (Supelco)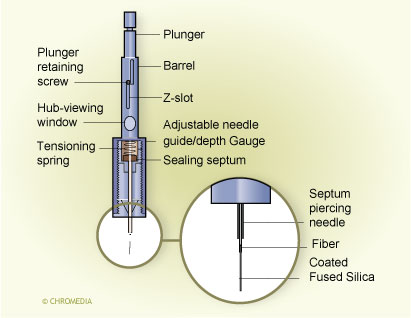 Fibre SPME can be performed in three basic modes: as direct extraction, in a headspace configuration, and in a membrane-protected approach (Fig.4).1 In direct extraction, the fibre coating is dipped into the aqueous sample and allows analytes to partition between the coating and the matrix. In headspace extraction, the fibre is placed in the headspace above the aqueous matrix during extraction. Only volatile analytes are extracted, but the method is advantageous for samples with high molecular weight interferences. When a sample contains both nonvolatile target analytes and high molecular weight interfering compounds, such as humic acids or proteins, application of direct or headspace SPME is quite difficult. In such cases, use of restricted-access materials or membrane-protected SPME results in better reproducibility and accuracy.
Fibre SPME can be performed in three basic modes: as direct extraction, in a headspace configuration, and in a membrane-protected approach (Fig.4).1 In direct extraction, the fibre coating is dipped into the aqueous sample and allows analytes to partition between the coating and the matrix. In headspace extraction, the fibre is placed in the headspace above the aqueous matrix during extraction. Only volatile analytes are extracted, but the method is advantageous for samples with high molecular weight interferences. When a sample contains both nonvolatile target analytes and high molecular weight interfering compounds, such as humic acids or proteins, application of direct or headspace SPME is quite difficult. In such cases, use of restricted-access materials or membrane-protected SPME results in better reproducibility and accuracy.
Fibre SPME modes: (a) direct extraction, (b) headspace SPME,
(c) membrane-protected SPME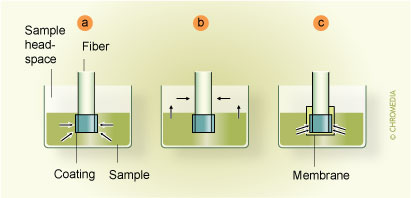 Fig. 4. Mode of fibre SPME operation: (a) direct extraction, (b) headspace SPME,
Fig. 4. Mode of fibre SPME operation: (a) direct extraction, (b) headspace SPME,
(c) membrane-protected SPME.
5. Modes of in-tube extraction:(a) passive (b) dynamic
 6. Different implementations of in-tube SPME (Click to enlarge)
6. Different implementations of in-tube SPME (Click to enlarge)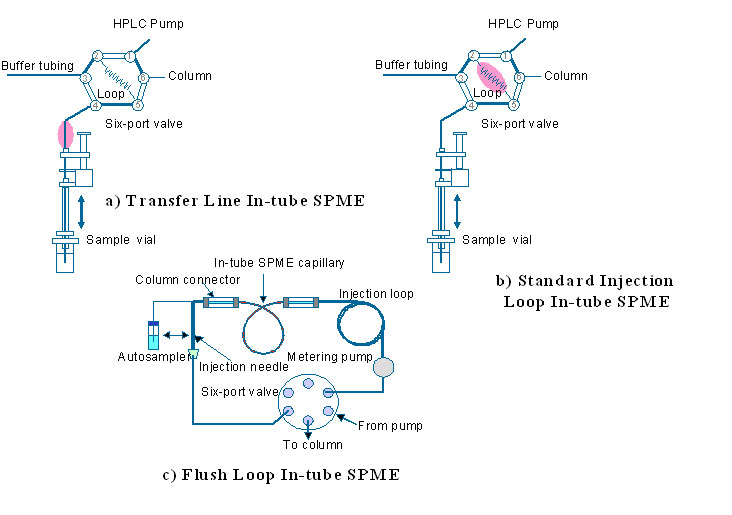 7. Configurations of SPME
7. Configurations of SPME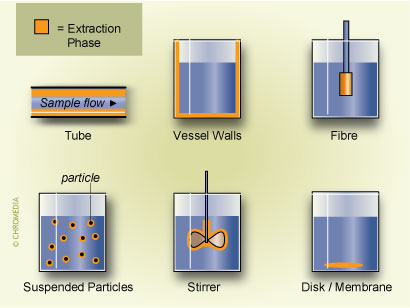 The extraction coating has also been used on other elements of the analytical system. Recently there have been reports of coating the interior of vessels,8 the exterior of magnetic stirring bars9 or even using pieces of poly(dimethyl)silioxane (PDMS) tubes and thin membranes10. Several of these implementations are shown in Fig.7
The extraction coating has also been used on other elements of the analytical system. Recently there have been reports of coating the interior of vessels,8 the exterior of magnetic stirring bars9 or even using pieces of poly(dimethyl)silioxane (PDMS) tubes and thin membranes10. Several of these implementations are shown in Fig.7 The main reason for developing these alternative approaches is to enhance sensitivity by using larger volume of the extraction phase (PDMS) and improving the kinetics of the mass transfer between sample and PDMS by increasing the surface to volume ratio of the extraction phase. The main disadvantage of these approaches, however, is loss of the convenience associated with a syringe configuration, in particular for the introduction of the sample into the analytical instrument.
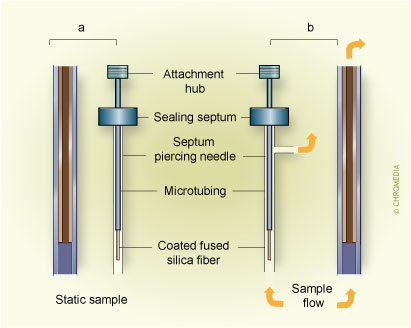
In addition to the analyte concentration measurement at a well-defined place in space and time, obtained by using the approaches discussed above, an integrated sampling is possible with a simple SPME system. This is particularly important in field measurements when changes of analyte concentration over time and place must often be taken into account.When the extracting phase is not exposed directly to the sample, but is contained in a protective tubing (needle) without any flow of the sample through it (Fig.8), the extraction occurs through the static gas phase present in the needle.
8. In-tube time weighted average sampling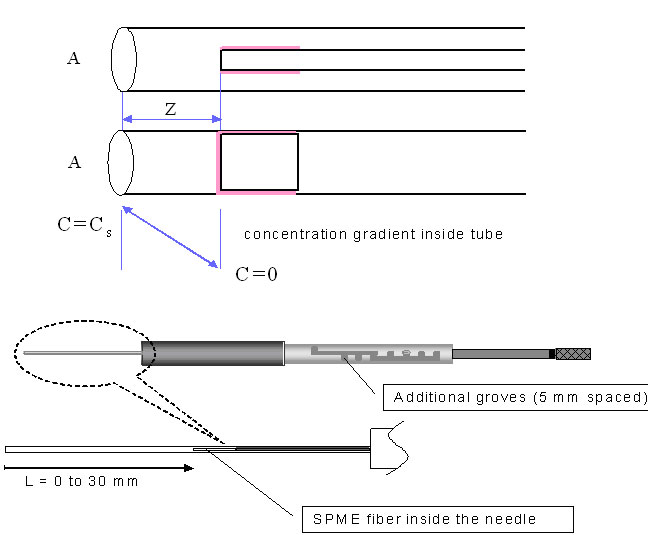 Fig.8. Use of SPME for in-tube time weighted average sampling. a) schematic, b) adaptation of commercial SPME manual extraction holder
Fig.8. Use of SPME for in-tube time weighted average sampling. a) schematic, b) adaptation of commercial SPME manual extraction holder
The integrating system can consist of extraction phase coating the interior of the tubing, or it can be an externally coated fibre withdrawn into the needle. These geometric arrangements represent a very powerful method able to generate a response proportional to the integral of the analyte concentration over time and space (when the needle is moved through the space)11. In these cases, the only mechanism of analyte transport to the extracting phase is diffusion through the gaseous phase contained in the tubing. During this process, a linear concentration profile (shown in Fig.8 A) is established in the tubing between the small needle opening, characterized by surface area A and the distance Z between the needle opening, and the position of the extracting phase. The amount of analyte extracted, dn, during time interval, dt, can be calculated by considering Fick's first law of diffusion:11
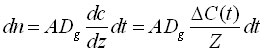 (7)
(7)
where ΔC(t)/Z is a value of the gradient established in the needle between needle opening and the position of the extracting phase, Z; ΔC(t) = C(t)-Cz, where C(t) is a time dependent concentration of analyte in the sample in the vicinity of the needle opening, and CZ is the concentration of the analyte in the gas phase in the vicinity of the coating. CZ is close to zero for a high coating/gas distribution constant capacity, then: ΔC(t) = C(t). The concentration of analyte at the coating position in the needle, CZ, will increase with integration time, but it will be kept low compared to the sample concentration because of the presence of the sorbing coating. Therefore, the accumulated amount over time can be calculated as:
![]() (8)
(8)
As expected, the extracted amount of analyte is proportional to the integral of the sample concentration over time, the diffusion coefficient of the analytes in the gaseous phase, Dg , the area of the needle opening, A, and inversely proportional to the distance of the coating position in respect of the needle opening, Z. It should be emphasized that equation 5 is valid only in a situation where the amount of analyte extracted onto the sorbent is a small fraction (below RSD of the measurement, typically 5%) of the equilibrium amount in respect to the lowest concentration in the sample. To extend integration times, the coating can be placed further into the needle (larger Z), the opening of the needle can be reduced by placing an additional orifice (smaller A), or a higher capacity sorbent can be used. The first two solutions will result in a lower measurement sensitivity. An increase of sorbent capacity presents a more attractive opportunity. It can be achieved by either increasing the volume of the coating, or its affinity towards the analyte. An increase of the coating volume will require an increase of the device size. The optimum approach to increased integration time, is to use sorbents characterized by large coating/gas distribution constants.
This approach has been implemented to date with the conventional fiber assembly, by retracting the fiber a known distance inside the needle (Figure 8B)1. The small size of the needle orifice limits diffusion to the sorbent surface, and the ultimate diffusion rate is also a function of the distance between the fiber tip and the end of the needle. Depending on the volatility and concentration of the analyte of interest, the fiber may be positioned either closer to or further from the end of the needle, to achieve the desired degree of non-equilibrium extraction and sensitivity. It would also be possible to implement this type of sampling with the sorbent coated on the interior wall of a capillary. To date however, the retractable needle implementation has gained the most attention, due to its ease of use and adjustability for the analyte and sample at hand.





