Abstract The mobile phase is by far the most important component of the HPLC system. Its composition plays a major part in obtaining good and reproducible results. The mobile phase can als cause a large number of problems if not enough attention is paid to its quality and use.
LevelBasic
Almost every application in HPLC demands the use of solvents of a very high purity.
Often "HPLC‑grade" solvents have to be used which are of a higher purity than "analytical grade". Special attention is paid to their water content, spectroscopic adsorption, solid contamination, and by‑products. These high standards are necessary to prevent several problems:
- Damage of pumps and blockages
- Lack in reproducibility
- Change in chromatographic behaviour
- Poor detector sensitivity (especially at low UV wavelength
- Noise and base‑line drift
- Extra peaks during gradient elution
Criteria for HPLC solvents:
- Compatible with the column
- Compatible with the detector
- UV wavelength cut-off
- Volatility (LC-MS)
 Purity
Purity - Viscosity
- Safety
- Price
Some impurities - "contaminations" - are added to the solvent by the manufacturer on purpose. They function as stabilizers to prevent the formation of by‑products (e.g. explosive peroxides). In general, stabilizers enhance the tenability (and lifetime) of the solvent.
Non‑stabilized solvents need to be replaced regularly which of course makes the system less cost effective. Well‑known examples of stabilized solvents are:
- Up to 1% methanol or ethanol in chloroform
- Butylated hydroxytoluene in THF
- Pentene in dichloromethane
Unfortunately the nature and quantity of these additives are not always specified on the label.
However, additives can have a few drawbacks. They can effect the retention behaviour as well as increase the UV background of the mobile phase.
The purity of water used in reversed phase systems (RPLC) is of particular importance because it has a very low elution strength. This means that when mobile phases are used with a high water content, the organic impurities in the water do not ![]() elute from the column but are retained in the top of the column.
elute from the column but are retained in the top of the column.
RPLC gradient analysis H2O->MeOH =0-100% Normal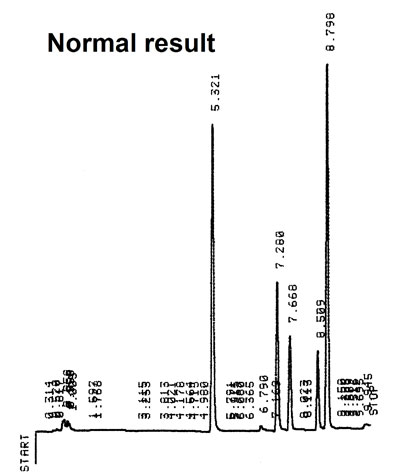
RPLC gradient analysis H2O->MeOH =0-100% What's this?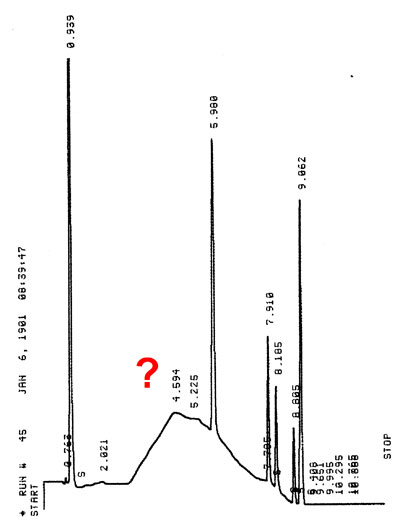
Poor water quality in gradient elution will result in strong baseline efects, ghost peaks, drift of the baseline (positive or negative) and instability of retention times after series of injections. Only HPLC grade water can be used. A HPLC water purification system is a good aternative.
Blank water-modifier gradient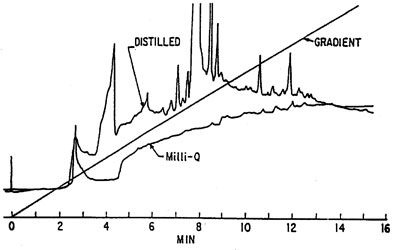 Dolan, Snyder. Troubleshooting LC-systyems. Humana Press.Tap‑ and demi-water are generally not suitable for use in RPLC and certainly not in combination with sensitive analyses and gradient elution. HPLC‑grade water is sufficient for most purposes but is quite expensive and pick up impurities from the atmosphere (e.g. organic vapours in the lab, cigarette smoke) once the bottle is opened.
Dolan, Snyder. Troubleshooting LC-systyems. Humana Press.Tap‑ and demi-water are generally not suitable for use in RPLC and certainly not in combination with sensitive analyses and gradient elution. HPLC‑grade water is sufficient for most purposes but is quite expensive and pick up impurities from the atmosphere (e.g. organic vapours in the lab, cigarette smoke) once the bottle is opened.
(Double) distilled water is also not very suitable for many applications.
Properties of HPLC water:
- Low UV absorption
- Very low conductivity
- Little background with electro-chemical detection
Products of choise:
- Use of HPLC grade water
- Purification of water with special treatment systems (such as Milli-Q from Milipore)
Another problem associated with water is the development of microbiologic contaminants (bacteria, algae), in particular when buffer salts are added to the water. ![]() This spontaneous growth causes blocked filters and columns and malfunctioning pumps. Mixing the water with an organic component of the mobile phase (e.g. THF or acetonitrile) strongly reduces bacterial contaminations.
This spontaneous growth causes blocked filters and columns and malfunctioning pumps. Mixing the water with an organic component of the mobile phase (e.g. THF or acetonitrile) strongly reduces bacterial contaminations.
For isocratic analyses it is best to mix the mobile phase in advance.
Solvent mixtures are usually stated in as volume ![]() fractions (v/v) and not in fractions of weight or Molar ratios. The mixing conditions of the eluent should be recorded in detail in the method so that others can reproduce it accurately!
fractions (v/v) and not in fractions of weight or Molar ratios. The mixing conditions of the eluent should be recorded in detail in the method so that others can reproduce it accurately!
Solutions of buffers and additives are usually described in Mol/l of g/l.
- The mobile phase should always be as fresh prepared as possible and stored in colored glass bottles in a cool place.
- The use of plastic bottles is not recommended because of the presence of plasticizers.
- The eluent bottle should always be placed above the level of the pump inlet head to obtain siphon action which will create a light over pressure at the inlet check valve of the pump.
- The index 'Properties of solvents' (verwijzing!) used in liquid chromatography lists some important properties of liquids such as refractive index, the polarity parameter and the UV‑cut off wavelength.
- Below 230 nm an acetate buffer can no longer be used. The same applies to methanol below 220 nm. Only acetonitrile can be used in this region of the spectrum.
| Solvent cut-off | nm | Solvent cut-off nm | nm |
| Water | 180 | Hexane | 195 |
| Acetonitril | 190 | Cyclohexane | 200 |
| Methanol | 210 | Chloroform | 245 |
| Ethanol | 205 | Ethyl acetate | 260 |
| Isopropanol | 205 | Benzene | 280 |
| THF | 225 | Toluene | 285 |
| Acetone | 330 | Nitro ethane | 380 |
The mixing of methanol and water does not only present problems for gradient elution, but also in isocratic analyses:
- Heat development
- Air
 bubbles
bubbles - Volume contraction
- Increase of viscosity
Degassing before or after mixing is essential.
The density after mixing MeOH and H2O increases as the volume decreases: For instance 500 ml methanol + 500 ml water result in approx. 960 ml mixture. Therefore the method of preparation of a mixture will strongly affect the final composition.
Only mixing of 500 ml methanol with 500 ml water is correct:
Mixing MeOH-H2O = 60-40 (v/v)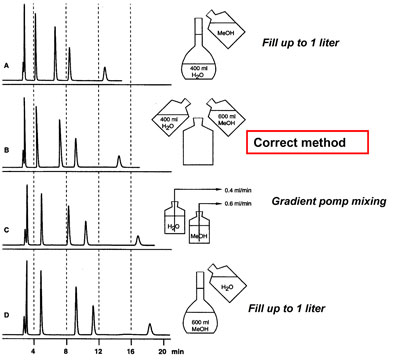
The viscosity of methanol/water mixtures varies widely and can reach very high values. Therefore a change in eluent composition will give ‑ at the same flow ‑ a different ![]() backpressure over the column. This factor has to be taken into account when considering the maximum pressure of the HPLC‑system. Viscosity water/modifiers
backpressure over the column. This factor has to be taken into account when considering the maximum pressure of the HPLC‑system. Viscosity water/modifiers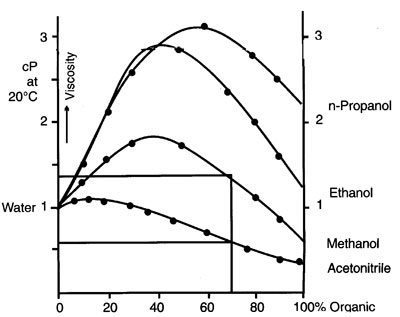 Figure 11
Figure 11
All previously discussed problems are less important for acetonitrile – water mixtures as mobile phase. Acetonitrile however, has its own drawbacks, such as volatility, toxicity and price.
The dissolved gases present in the various liquids must be removed before they are pumped through the chromatograph. When the eluent is insufficiently degassed, air and vapour bubbles will form inside the system causing several malfunctions.
The parts of the system most prone to those problems are:
- The pump
- The mixing chamber of the gradient system the detector
The problems associated with this are:
- Irregular or pulsating flow, or no flow at all
- Irregular eluent composition
- Spikes and base‑line “jumps” due to bubbles inside the detector or a very high background signal, noise and drift (especially fluorescence and electrochemical detection)
Degassing mobile phase mixtures should be done after the several components are mixed. The separate liquids can contain more gas than the mixture. In gradient system each solvent is (automatically) degassed before it reaches the mixing chamber or the pump.
Several techniques can be used to degas the eluent:
- Vacuum degassing (on-line in many HPLC systems)
- Vacuum filtration
- Ultrasonic vibration
- Purging with helium
Efficiency of solvent degassing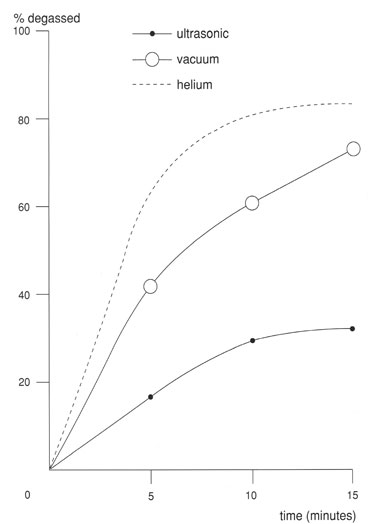
Degassing and fluorescence detection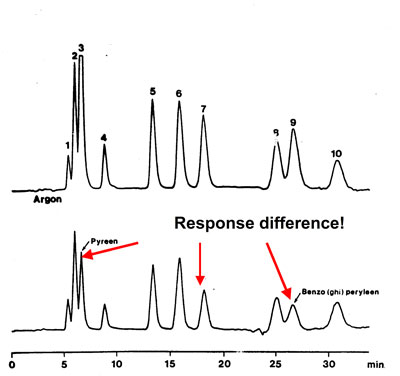
Degassing by bubbling helium through the different solvents is most effective, but takes quite some (pure!) helium. By far the most efficient method is the helium method, in which the eluent is purged with small helium bubbles. Helium has a very low solubility in most liquids used. Oxygen, CO2 and nitrogen diffuse into the helium bubbles and are thus removed from the eluent. This method is very efficient and the quickest of all methods. In gradient elution systems, in which 2 or more pure solvents are mixed, a continuous stream of helium can be bubbled through each of the solvents to degas them effectively. If the mixture contains a very volatile component the purge time has to be standardized to reproduce selective evaporation.
In-line degassers are common now, which are an integrated part of the HPLC system between the solvent reservoirs and the mixing unit of the gradient pump. These units are equipped with semi-permeable tubing, where the solvent is flowing through (one capillary for each bottle). Outside these tubes a vacuum is generated by a pump. As the wall of the capillary is diffusive for gases and not for solvent molecules, dissolved gasses are drawn from the flowing solvents, resulting in adequate degassing for normal (gradient Degassing by bubbling helium ) applications.
Filtering with a vacuum is an elegant degassing method as the vacuum and the filter (0.2 or 0.5 μm) does not only take care of degassing but also of purifying the eluent. As in the case with degassing solely with a vacuum, the main problem is selective evaporation of one or more of the solvents.
Degassing by ultrasonic vibration is not as good a method compared to the others and bares the risk of changing the mobile phase compositions as a result of selective evaporation of the more volatile parts of the solvent mixtures.
For fluorescence and electrochemical detection methods the removal of oxygen from the eluent is of the utmost importance because oxidation of the sample makes the detection less sensitive. For these detectors the helium method is again the most effective one.
In many of the "reversed phase" analyses of strongly polar or ionic substances the aqueous mobile phase is buffered to control the pH of the eluent. This is essential for stable chromatographic characteristics and good peakshape of acids and basic compounds.
The mentioned pH value in the method for the mobile phase is specified for the aqueous solution of the buffer only. The addition of an organic modifier influences the pH of the total solvent. For a buffer‑ methanol mixture this means that the proton activity depends on the composition of the mixture. As a result of this the pK*‑value (and thus the pH* value of the buffer‑methanol mixture) changes during a gradient run.
Salt additives:
- Poor solubility of buffer salts in organic modifiers
- pH of the mobile phase is specified for the water fraction, not the mixture
- After use flush the complete system with water/ modifier
- Use fresh buffers < 1 week) only
The concentrations of the buffers used depend on the application (and of course on the pH).
- As the amounts of injected ionic substances are usually very small, the buffer concentrations do not have to exceed 0.05 M for most separations.
- High capacity ion exchangers sometimes need buffer concentrations over 0.25 M as not only the pH is of importance but also the concentration of counter ion.
- In Gel Permeation Chromatography an aqueous mobile phase is sometimes used with salt concentration of up to 6 M (urea, guanidines) are not uncommon. Those concentrations are necessary to elute proteins by means of a "salting out effect". In this way the interaction between the gel and the sample is strongly minimized.
The use of salts and other additives in the eluent can cause several problems that need special attention:
- Reduced solubility of the organic modifier in a buffer solution
- Corrosion of the pump and other parts of the system
- Channels in the column packing
- Salting out during stop‑flow conditions
- Bacterial growth in storage of buffer solutions
When an acid or a base is added to a salt solution in order to reach a desired pH, there is a risk that the salt is precipitating when the organic modifier is added. Usually settling out or crystallization of the salt is taking place when the eluent becomes cloudy after (gradient) mixing. In particular the solubility of phosphate buffers in acetonitrile is poor (< 50 mM phosphate / l in > 50% ACN).
Low buffer concentration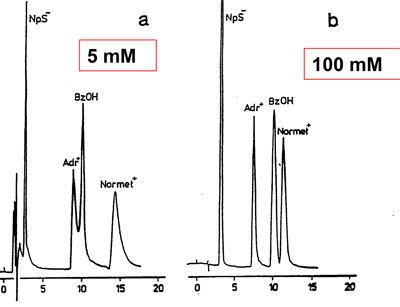
Effect of buffer type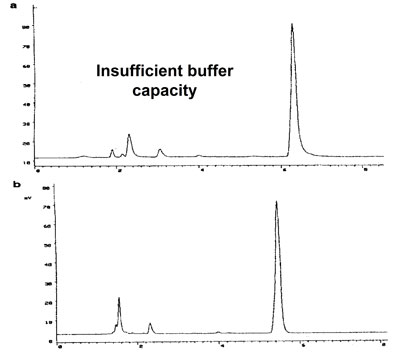
a. 5 mM phosphate buffer @ pH = 4.6. Insufficient buffer capacity: the pKa value of phasphate is 7.0
b. 5 mM acetate buffer @ pH = 4.6 Correct application of the acetate buffer: pKa = 4.5
Gradient elution in particular is subject to the situations described above. If there is any doubt, always prepare a test solution with the desired end composition of the eluent to ![]() check whether settling out or crystallizing occurs.
check whether settling out or crystallizing occurs.
To avoid this, keep the pump running overnight at a low flow rate (recirculate the solvent in case of isocratic conditions with pre-mixed solvents).
The separation of strongly polar substances is done with eluents to which special modifiers are added to reduce the silanol activity of the packing (like TEA, triethylamine). As a result the retention and tailing, especially of nitrogen‑containing compounds is thereby reduced.
To facilitate the separation of ionic substances on reversed phase materials it is necessary to add so‑called ion‑pair reagents to the mobile phase. Examples of such ![]() reagents are alkylsulfonic acids for cation separations and trimethyl- alkylammonium salts for
reagents are alkylsulfonic acids for cation separations and trimethyl- alkylammonium salts for ![]() anions. Column conditioning for Ion Pair Chromatography
anions. Column conditioning for Ion Pair Chromatography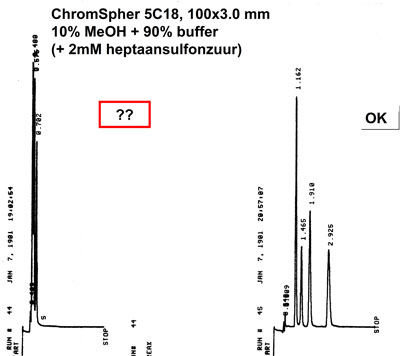
Left chromatogram is the result of a poor equilibration of the column with this highly aqueous mobile phase. Chain collapse (insufficient conditioning of the C18 chains of the packing with the mobile phase) results in a very poor retention of the complete chromatogram. Flushing the column first with > 30% methanol, followed by proper equilibration with the mobile phase (including the ion pair reagent heptanesulphnic acid) with bring the column back to it's original quality.
The simplest way to change the mobile phase composition is done in gradient pump systems. Most instruments are equipped with a high pressure 3‑way valve (purge valve) between pump and injection valve. This valve makes it possible to flush the system at a high flow rate between different solvents.
When changing the eluent the following points should be considered:
- Complete miscibility of both old and new mobile phase is required
- Do not introduce air bubbles in the inlet tube of the pump
- Maximum pressure of the system (open the purge valve completely!)
Saving eluent
The expense of solvents can be limited to some extent by a careful choice of solvent (see elsewhere in this chapter). The amount of solvent use can be reduced as follows:
- Recycling. This is a suitable technique for routine analyses at not too high detector sensitivities and sample concentrations. This method is of course limited to isocratic pre-mixed mobile phases only.
- Smaller column diameter. The flow rate through the column is reduced in proportion to the square of the column diameter at the same linear velocity. This will save solvent but does not save time. However, for internal diameters below 2 mm special equipment is required.
- Good planning and organization. Never make more solvent than necessary. A lot of mobile phases are disposed because of pollution, bad labelling and degradation.
Although this subject does not directly fall within the scope of Trouble Shooting, it is important enough to pay some attention to it. HPLC operates with solvents which are often harmful and so special care should be taken regarding use and storage of such solvents.
Toxicity. Although most solvents are partly aqueous they still contain a considerable amount of organic solvent. Many of these are hazardous even at very low concentrations. Acetonitrile, for example, has a MAC‑value of 40 ppm which makes it one of the most poisonous solvents used in HPLC. Always bear in mind however that toxicity is linked to the handling of different volumes of solvents.
THERE ARE NO TOXIC SUBSTANCES, ONLY TOXIC QUANTITIES
This implicates that the danger connected with solvents can be limited by avoiding any contact with solvents and vapours (fume hoods, laboratory ventilation).
Flammability. Most organic solvents are, due to their volatility and reactivity with oxygen, very flammable. A few precautionary measures should be taken if one wants to work safely with HPLC solvents:
- Storage of large quantities should be carried out in separate, purpose designed, fireproof and well ventilated spaces and cabinets.
- Storage of opened bottles or small quantities in a well ventilated, fire resistant fume hoods.
- Old supplies or used solvents should be removed regularly
- Mixing, degassing and changing of solvents should take place in a fume hood.
- Use of properly labelled waste bottles
- Observe the usual safety precautions in laboratories such as prohibition to smoke, protective clothing, wear spectacles, etc.
Broad peak in gradient chromatogram figure 22
figure 22





