Abstract A number of separation systems can be used to determine organic compounds in complex matrices. The most important techniques are LC, GC and CE. In order to develop an adequate analytical method / procedure, the combination of SP/ST, separation and the reaction/detection procedure is more critical than the separation mode itself. To choose the best option from the large number of possibilities you need clear guidelines. The most suitable method can be found by answering the questions given in the following tables. These questions are related to the physico-chemical properties of the analyte and of the matrix as well as the objectives of the overall method.
LevelBasic
- The chemical structure of the analyte and the physico-chemical properties of the matrix together with the desired selectivity and sensitivity determine the most suitable isolation and quantitation system.
- The required clean-up procedure strongly depends on the chosen LC–detection combination. In the case of a selective detection technique isolation of the analyte from the matrix may be superfluous, but this can almost never be achieved.
- With the variety of currently available LC-detection systems it is not always possible to obtain the desired detectability, with the necessary precision, without a chemical manipulation – derivatization – of the analyte.
We must realize that at the moment the goal of the analysis has been identified all the available options must be reviewed with respect to accuracy, precision, cost, etc. The amount of labour, time required and the degree of automation also are important aspects. This chapter on method development provides the necessary know-how to do a good job in this respect.
Bio-analysis
Accurate, sensitive and selective (bio-)analytical determination methods for drugs and drug products (e.g. degradation products, metabolites) are of major importance in a number of bio-analytical research fields: therapeutic-drug monitoring (TDM), metabolic profiling, toxicology, drug pharmacology, and pharmacokinetics and stability. The concentration of drugs and metabolites in whole blood, plasma, urine, saliva or tissue homogenates is in many cases in the nanogram (ng/mL) or even in the picogram (pg/mL) range, necessitating the use of selective and sensitive separation and detection methods after isolation and cleaning of the analyte from the excess of endogenous and exogenous components in the biological matrix.
Chromatographic separation techniques such as LC in combination with selective and sensitive (reaction)/detection devices have gained an enormous popularity in determining compounds of biomedical interest. Important reasons are that for newly developed drugs:
- The potency is higher compared with analogues developed about ten years ago;
- The therapeutic window – difference between the minimum therapeutic concentration and the toxic concentration – for some of these drugs is rather small;
- The risk of severe side-effects is increasing;
- Accumulation by repetitive dosage may be a problem;
- A wide individual variance may require additional precautions.
As a result TDM requires still more sophisticated techniques. In LC, therefore, there is a trend to use columns with smaller internal diameters (I.D.), more efficient stationary phases using smaller particle sizes, and new more selective sorbents.
Apart from the classical methods, such as ![]() titrimetric and gravimetric techniques, many instrumental techniques have been developed, for the determination of not only the active ingredient, but also the quantification of related compounds or impurities associated with it. The recently developed analytical methods have the advantage of not only using small amounts of sample, reagents and less time, but also produce accurate results. These analytical techniques could either be:
titrimetric and gravimetric techniques, many instrumental techniques have been developed, for the determination of not only the active ingredient, but also the quantification of related compounds or impurities associated with it. The recently developed analytical methods have the advantage of not only using small amounts of sample, reagents and less time, but also produce accurate results. These analytical techniques could either be:
- Physico-chemical methods. Spectroscopy, including colorimetry and spectrophotometry covering ultra-violet and visible region or fluorimetry,
 nephelometry or turbidimetry, and nuclear-magnetic resonance and mass spectrometry
nephelometry or turbidimetry, and nuclear-magnetic resonance and mass spectrometry - Electro-analytical methods. The electro-analytical methods cover potentiometry, amperometry, voltammetry, electrophoresis and polarography.
- Separation-based methods. (High-performance) liquid chromatography (LC), (high-performance) thin-layer chromatography (HPTLC / TLC),
 capillary electrochromatography (CEC), supercritical-fluid chromatography (SFC) and gas chromatography (GC).
capillary electrochromatography (CEC), supercritical-fluid chromatography (SFC) and gas chromatography (GC). - In addition to chromatographic separation methods, electrophoretic techniques, like capillary electrophoresis (CE),
 isotachophoresis (ITP), gel electrophoresis (GE) and isoelectric focusing (IEF), are relatively popular for the separation and quantitation of (charged) organic compounds.
isotachophoresis (ITP), gel electrophoresis (GE) and isoelectric focusing (IEF), are relatively popular for the separation and quantitation of (charged) organic compounds.
Drug analysis
The still increasing interest on the determination of drugs has forced the analytical chemists to develop methods for their trace ![]() analysis in the presence of biological matrices. These methods should be rapid, precise, accurate and cost effective. Although costly sophisticated instruments like LC, HPTLC, GC, GC-MS and LC-MS are available, the spectrophotometer is being preferred by ordinary laboratories for its simple, economical and easy handling techniques. An overview:
analysis in the presence of biological matrices. These methods should be rapid, precise, accurate and cost effective. Although costly sophisticated instruments like LC, HPTLC, GC, GC-MS and LC-MS are available, the spectrophotometer is being preferred by ordinary laboratories for its simple, economical and easy handling techniques. An overview:
| Separation techniques: | Acronym | |
| Liquid chromatography | LC | |
| (Capillary) gas chromatography | GC | |
| Supercritical-fluid chromatography | SFC | |
| Thin-layer chromatography | TLC | |
| Capillary (zone) electrophoresis | C(Z)E | |
| Hydrophobic-interaction chromatography | HIC | |
| Immunoaffinity chromatography | IAC | |
| Radio-chromatography | RC | |
| Size-exclusion chromatography | SEC | |
| Spectroscopic techniques: | ||
| Mass spectrometry | MS | |
| Ultraviolet-visible absorption | UV-VIS | |
| Emission spectrometry | ||
| Fluorescence | FL | |
| Phosphorescence | P | |
| Chemiluminescence | CL | |
| Colorimetry | ||
| Fourier-transform infrared | FTIR | |
| Nuclear magnetic resonance | NMR | |
| Electro analytical techniques: | ||
| Amperometry | AMP | |
| Coulometry / Potentiometry / | ||
| Polarography / Voltammetry | ||
| Bio-assay techniques: | ||
| Radioimmunoassay | RIA | |
| Immunoradiometric assay | IRMA | |
| Enzyme-multiplied immunoassay | EMIT | |
| Substrate-labeled fluorescent immunoassay | SLFIA | |
| Enzyme-linked immunsorbentassay | ELISA | |
| Receptor assay | ||
| Protein-binding assay | ||
| Enzymatic assay | ||
| Microbiological assay | ||
Guidelines for method choice in analytical chemistry
The necessary sampling and SP steps for the determination of analytes in complex and dirty matrices is one of the most troublesome to perform, and therefore, the degree of SP depends on quite a number of parameters. The most important ones are the:
- Concentration of the analyte;
- Composition of the matrix;
- Number of samples to be analyzed;
- Chosen separation / detection system.
This paragraph presents general guidelines for separation method selection. To simplify the tables only organic molecules with a MW of less than 1 kD are taken into account. This means that some of the separation techniques mentioned earlier e.g. SEC are not taken into consideration. (HP)TLC means that in case quantitative or semi-quantitative results should be obtained high-performance TLC (HPTLC) should be used instead of classic TLC. The abbreviations used are explained in the 'List of abbreviations and glossary of symbols'.
Tables 1, 2, 3: Guidelines for method choice in analytical chemistry.
| Questions to be answered: | Possible techniques: | |
| Aggregation phase: | Gas | GC, SFC |
| Liquid / solid | CE, GC, IEC, LC, OPTLC, (HP)TLC, SFC | |
| Charge: | Not present | GC, LC, MECC, OPTLC, (HP)TLC, SFC |
| Present | CE, IEC, LC (IP, IS) | |
| Functional groups: | Not present | Almost no derivatization possibilities |
| Present | Derivatization potential | |
| Polarity: | Low | Non-polar sorbents in chromatography |
| High | Polar sorbents in chromatography | |
| Saturations (aromaticy): | Aliphatic | AMP, CL, CON, ECD, FID, FS, IR, NMR, NPD, PID, POL, RI, SIM, |
| Conjugated / aromatic | AMP, CIF, CL, CON, ECD, FID, FS, IR, LIF, NMR, NPD, PID, POL, RI, SIM, UV-VIS | |
| Solubility: | Polar solvents | CE, IEC, LC, OPTLC, (HP) TLC |
| Non-polar solvents | GC, LC, OPTLC, (HP)TLC, SFC | |
| Volatility: | Low | CE, IEC, LC, OPTLC, (HP)TLC, SFC |
| High | GC, LC, SFC |
| Complexity of the matrix: | Degree of automation, amount of effort: | |
| Analyte - matrix binding: | None | No special precautions |
| Yes | Denaturation procedures should be used in case of drug -protein binding or other analyte - matrix disrupting techniques | |
| Minimum detectable concentration(s): | 1 - 1000 μg/mL | CE, GC, LC, OPTLC, (HP)TLC, SFC, AMP, CIF, CON, ECD, FID, IR, NMR, NPD, PID, POL, RI, SIM, UV-VIS |
| 1 - 1000 ng/mL | CE, GC, LC, OPTLC, HPTLC, SFC AMP, CIF, CL, ECD, LIF, NPD, PID, SIM, UV-VIS | |
| 1 - 1000 pg/mL | CE, GC, LC, SFC, AMP, CIF, CL, ECD, LIF, SIM | |
| 1 - 1000 fg/ml | CE, LC, (SFC), CL, LIF | |
| Stability: | Bad | Stabilizing procedures |
| Good | No stabilizing procedures needed |
| Availability equipment: | Determines choice of SP / ST, separation / detection system | |
| Available expertise: | Determines choice of system components and the degree of automation | |
| Number of solutes to be determined | < 10 | CE, GC, LC, OPTLC, SFC |
| > 10 | CE, GC, LC | |
| Number of samples to be analyzed in each series: | Degree of automation | |
| Number of sample series to be analyzed | Degree of automation | |
| Profiling of analytes: | Yes | CL-MS/(MS), GC-MS/(MS), LC-FTIR, LC, NMR, LC-DAD |
| No | No restriction in separation / detection mode | |
| Rationale for analysis | Qualitative | CE, GC, LC, OPTLC, SFC, TLC |
| Semi-quantitative | CE, GC, LC, OPTLC, SFC, TLC | |
| Quantitative | CE, GC, LC, SFC | |
| Reason for analysis: | Legal | Reliability most important parameter |
| Toxicological | Speed most important parameter | |
| TDM | Throughput important parameter | |
| Drug development | Screening and identification of metabolites important parameters | |
| Ruggedness of the method | High | CE-DAD, GC-ECD, GC-FID, GC-NPD, LC-CIF, LC-DAD, automated reaction / detection systems |
| Low | No restrictions | |
In analytical chemistry most of the time selectivity, sensitivity, reproducibility, speed and costs are the critical parameters when organic compounds must be determined in complex samples. This means that to develop a qualitative or quantitative method, we will select an optimal combination of the following techniques:
| Sample preparation / treatment | Labeling | Separation | Labeling | Detection |
| Precipitation | Pre-column | TLC | Post-column | UV-VIS |
| LLE |
| HPTLC |
| FL |
| SPE |
| OPLC |
| AMP |
| Column switching |
| SFC |
| CL |
| Dialysis |
| HIC |
| LIF |
| Dehydration |
| AC |
| MS |
| Distillation |
| SEC |
| IR |
| Electrophoresis |
| GC |
| NMR |
| Freezing |
| LC |
| FID |
| Hydrolysis |
| CE |
| RI |
| Micelles |
|
|
| NPD |
| Immunoaffinity |
|
|
| POL |
| Lyophilization |
|
|
| ECD |
| Microwaves |
|
|
| AAS |
| Soxhlet |
|
|
| AES |
| Ultrafiltration |
|
|
| PID |
| SFE |
|
|
| CON |
| Saponification |
|
|
| DAD |
In order to choose the proper methods and to develop a suitable method first of all, we have to answer the principal questions in the "Guideline tables 1, 2 and 3". After answering these questions first of all the separation and detection technique must be chosen, and based on this the sampling and SP procedure can be selected.
From the tables it can be seen that numerous combinations of separation and detection techniques can be chosen for a particular problem.
- The first choice that should be made is which mode of chromatography/electrophoresis should be applied. In general, the analyte(s) should be determined quantitatively, which means that selectivity and sensitivity are the most critical parameters.
- We see from the table 3 that CE, GC and LC will be the most obvious separation techniques.
- The - most likely - suitable detection modes are UV-VIS, DAD, FL and MS in combination with LC, FID, ECD, NPD and MS for GC, and UV-VIS, DAD, LIF and MS in combination wit CE. Especially, the use of MS or MS/MS approaches is still gaining popularity.
- The sampling and SP approach will be selected on the basis of the chosen separation-detection system. Normally a combination of initial, non-selective, technique in combination with a selective SP procedure will be applied.
The physico-chemical properties of the analytes and the origin of the sample (e.g. biological, environmental, food) determine which separation technique can be used. Some important features:
- Using (capillary) GC mainly volatile solutes, compounds that are thermally sufficiently stable, and have a molecular-weight of less than 500 can be determined without a derivatization procedure.
- An important feature is that the separation power of GC is about 100 times higher as of LC. This means that if a problem can be solved with both LC and GC, GC normally is the first option.
- Other differences between GC and LC are that automated sample preparation is LC is more sophisticated than in GC, but in GC a number of selective detection approaches are available, meaning that the requirements for the sample preparation are less critical. Nowadays, both LC-MS(/MS) and GC-MS(/MS) techniques can be used for routine analysis.
- Another limitation of GC, the relatively small injection volumes, can be circumvented by using PTV (Programmed Temperature Vaporizer) injectors. The result is that volumes up to about 1 mL can be injected, even in a narrow-bore capillary GC system.
- CE is in particular developed for the separation of charged compounds. CE is mainly suitable for the qualitative and semi-quantitative determination of relatively high concentrations of organic and inorganic compounds in relatively simple matrices.
LC is a technique that can be used for the separation of polar and non-polar compounds, for charged and neutral compounds, and for volatile and non-volatile compounds.
HPLC principle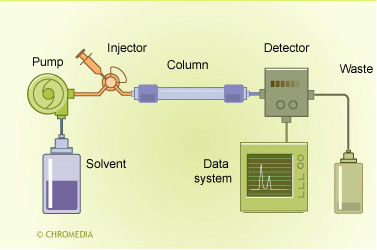
In particular, when mixtures of compounds with different polarities, i.e., drug and their metabolites, parent compounds and their degradation products, should be measured, LC is the method of choice. An overview on the selection of the phase systems can be found in the table:
| Analytes soluble in aqueous solvents | Ionic (acidic, basic, amphoteric) |
|
|
| LC | RP-IS | RP-IP | IE |
| Sorbent | Non-polar | Non-polar | Polar |
| Eluent | Modifier / Buffer | Modifier / Buffer + IP reagent | Buffer |
| Analytes soluble in aqueous solvents | Non-ionic |
|
|
| LC | RP | Partition |
|
| Sorbent | Non-polar | Polar |
|
| Eluent | Modifier / Buffer | Modifier / Buffer |
|
| Analytes soluble in organic solvents | Ionic (acidic basic, amphoteric) |
|
|
| LC | RP-IS | RP-IP | Partition |
| Sorbent | Non-polar | Non-polar | Polar |
| Eluent | Modifier / Buffer | Modifier / Buffer + | Modifier / Buffer |
| Analytes soluble in organic solvents | Non-ionic |
|
|
| LC | RP | Partition (Adsorption) |
|
| Sorbent | Non-Polar | Polar |
|
| Eluent | Modifier / Buffer | Organic solvents |
|
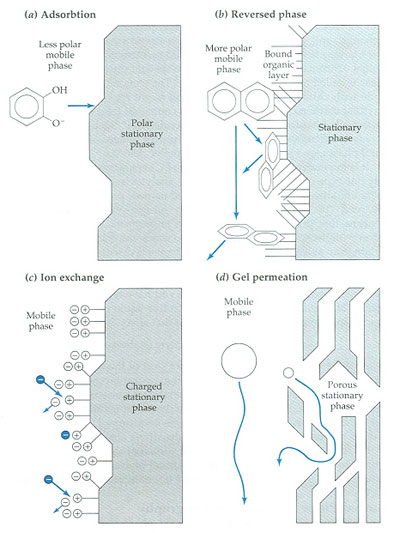
Different mechanisms of retardation in liquid chromatography (J.F. Rubinson, K.A. Rubinson, Contemporary Chemical Analysis, Prentice Hall, Upper Saddle River, 1998).
Adsorption LC
Adsorption LC probably is one of the oldest types of chromatography. It is using a mobile liquid or gaseous phase that is adsorbed onto the surface of a stationary solid phase. The equilibration between the mobile and stationary phase accounts for the separation of different solutes. In this mode a polar sorbent (i.e., silica, alumina) in combination with a non-polar eluent is used. The limited stability and reproducibility, as well as the bad peak shape which is frequently inherent with the analysis of neutral (basic) compounds explains its limited popularity. In most applications adsorption LC can be replaced by normal-phase (NP) partition chromatography.
Nowadays, adsorption chromatography has been replaced by partition chromatography. This form of chromatography is based on a thin film formed on the surface of a solid support by a liquid stationary phase. The solutes equilibrate between the mobile phase and the stationary liquid. Partition chromatography can be performed in the NP and in the reversed-phase (RP) mode.
Normal-phase LC
NP-LC is performed by using silica sorbents which or chemically modified with polar and/or hydrophilic functional groups and in combination with non-polar eluents. The most popular sorbents are the amino- and the cyanopropyl sorbent. Both sorbents can be used for the separation of neutral polar compounds. The cyanopropyl sorbent can be used in combination with both polar and non-polar eluents. The use of NP-LC can be advantageous in case of fluorescence detection. Adsorption chromatography (LSC)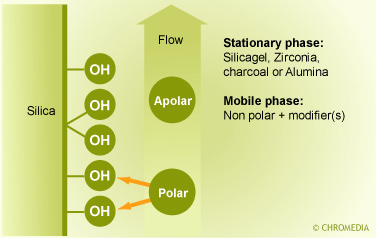
Reversed-phase LC
The most popular mode of LC is RP-LC. In this case a non-polar sorbent is combined with polar eluents. The positive features of RP-LC are a fast equilibration, excellent stability, good reproducibility and peak symmetry, as well as a large application area. For neutral compounds, in principle, RP-LC is the method of choice. The most popular sorbents are the cyanopropyl, octyl and octadecyl phases. In case a more hydrophobic sorbents or an extreme pH value (<2 or >8) must be applied, polymeric (i.e. copolymers of styrene-divinylbenzene) or graphitized carbon can be applied.
Ion-exchange LC
IEC is performed using either resin (polymeric sorbent) or silica gel material which are chemically modified with charged functionalities (Figure below). These functionalities are covalently attached to the sorbent. Solute ions of the opposite charge in the eluent are attracted to the sorbent by means of electrostatic forces. IEC is applied for the separation of ionized compounds. Depending of the ion-exchanger, weak or strong, the retention is controlled by the ionic strength or the pH, respectively.
Principle of ion-exchange chromatography 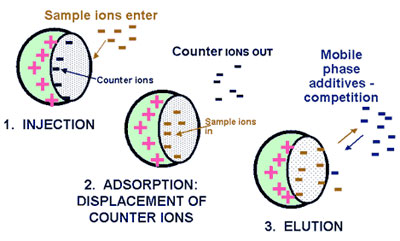 www.forumsci.co.il
www.forumsci.co.il
Ion-pair LC
In IP-LC charged counter-ions are used in combination with a neutral, normally hydrophobic, sorbent. IP-LC can be applied as an alternative for IEC, for the separation of charged compounds. The disadvantage of IP-LC is that, depending on the counter-ion used, equilibration is rather time-consuming.
Ion-suppression LC
IS-LC is a special mode of RP-LC in which the pH of the eluent is adapted in such a way that the ionization of the analytes is suppressed. This approach is mainly used for the separation of weak acids.
An overview of separation techniques: 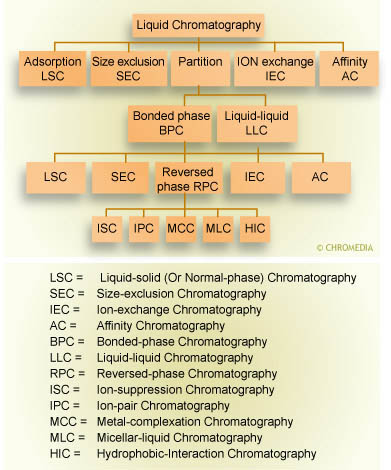
One of the limitations of LC may that at the moment no universal and sufficiently sensitive detectors are available; this in spite of the fact that a wide variety of detection systems varying from selective (amperometric) to universal (refractive index) and from insensitive (conductometry) to sensitive (fluorescence) are available. The limited applicability of selective detectors can be overcome by labeling the analyte with a suitable reagent.
A derivatization procedure can be part of the SP procedure, and can be performed before and after the chromatographic / electrophoretic procedure.
UV-VIS
Fixed-wavelength absorption detectors (UV-VIS) are in particularly suitable for the quantitation of one or more compounds possessing the same or similar chromophores. The limitation is that identification cannot be performed. Diode-array detectors (DAD) can be used to obtain some structural information, while MS techniques can be used to perform structure confirmation of analyte(s) (Figure below).
Diode Array Detector UV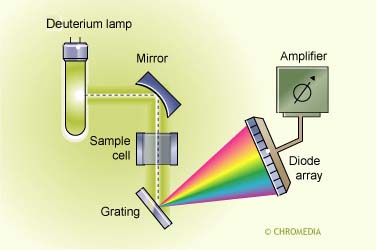
 Diode-array spectrum (www.ribbonchem.it).
Diode-array spectrum (www.ribbonchem.it).
The choice of the detection system mainly depends on the required limit of determination and the structure of the analyte. For determination limits in the μg/mL – high ng/mL range, UV-VIS detection can be used. Unsaturated compounds can be detected at wavelengths over 250 nm, while aliphatic compounds can be detected between 200 and 250 nm. The application of wavelengths below 280 nm is not favourable, in many cases, because of matrix interferences. However, in combination with a proper choice of eluent and SP procedure, wavelengths between 200 and 210 nm can be used to improve the sensitivity.
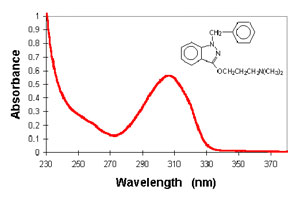 Absorption spectrum of benzydamine (photobiology.com).
Absorption spectrum of benzydamine (photobiology.com).
The absorption maximum not always is the optimum detection wavelength, because the background signal also can be relatively high at this wavelength. In general it can be stated that the matrix background will be lower at longer wavelengths, but at these wavelengths also the molar absorptivities of the analytes will be lower. This means that always a compromise should be found to determine the maximum signal-to-noise ratio. During method development, in order to find the optimum detection wavelength, the presence of a DAD spectrum can be rather helpful. A proper optimization can result in a gain in sensitivity of 1-2 orders of magnitude. Another way to increase the sensitivity is to apply a proper SP procedure, to remove interfering matrix components.
In case sensitivities in the ng/mL to high pg/mL are required, UV-VIS detection can be used for conjugated and aromatic compounds, while for aliphatic compounds a more sensitive technique like AMP, FL or mass MS should be used:
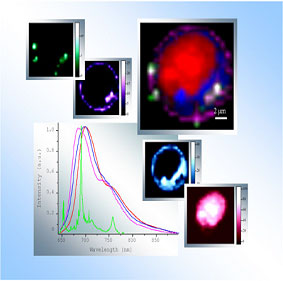 Simultaneous detection of SERRS and fluorescence of anticancer drug mitoxantrone within a single living cancer cell (MCF-7) by confocal spectral imaging. Bottom left window shows the reference spectra of a) fluorescence of drug-DNA complex (red); the drug fluorescence in high and low polarity environment (blue and violet, respectively); b) SERRS of the drug (green). Fitting the intracellular spectra with the reference ones provides the respective quantitative maps (monochrome images) that can be superposed (multicolor image) to analyze their colocalisation with cellular compartments: nucleus (red); polar cytosolic regions (blue); cellular membrane and those of organelles (violet). The SERRS map indicates the co-localization of silver colloid aggregates with the membranes (www.jobinyvon.com).
Simultaneous detection of SERRS and fluorescence of anticancer drug mitoxantrone within a single living cancer cell (MCF-7) by confocal spectral imaging. Bottom left window shows the reference spectra of a) fluorescence of drug-DNA complex (red); the drug fluorescence in high and low polarity environment (blue and violet, respectively); b) SERRS of the drug (green). Fitting the intracellular spectra with the reference ones provides the respective quantitative maps (monochrome images) that can be superposed (multicolor image) to analyze their colocalisation with cellular compartments: nucleus (red); polar cytosolic regions (blue); cellular membrane and those of organelles (violet). The SERRS map indicates the co-localization of silver colloid aggregates with the membranes (www.jobinyvon.com).
Amperometric (AMP) detection only can be used when oxidable or reducible structures are present in the molecule that can be oxidized at an acceptable potential. Electrochemical-active functional groups can be introduced into a molecule, during the SP procedure, via a derivatization procedure:
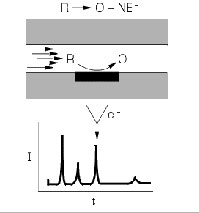 Amperometric detection in liquid chromatography (www.epsilon-web.net).
Amperometric detection in liquid chromatography (www.epsilon-web.net).
It is preferred to use a relatively low potential, which means that interferences by eluent and matrix components can be avoided. The LOQ values can be in the low pg/mL range.
Fluorescence (FL) detection
Using FL detection, determination limits in the low pg/mL range can be obtained. A derivatization procedure, using a suitable fluorescence probe, can be used to derivatize which do not possess native fluorescence. The derivatization reaction can be performed either before or after the separation.
Principle of a fluorescence detector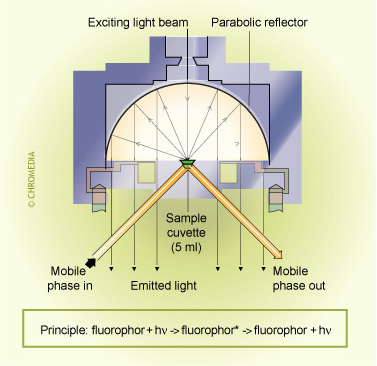
Applying laser-induced fluorescence (LIF) the detectability can be improved with a factor of 10 – 100. In spite of the improved sensitivity, LIF is still not rather popular because of the limited availability of commercial systems and the fact that only a few lasing wavelengths are available, which means that a derivatization procedure cannot be avoided in many cases. The introduction of relatively inexpensive diode lasers certainly will increase the applicability of LIF detection.
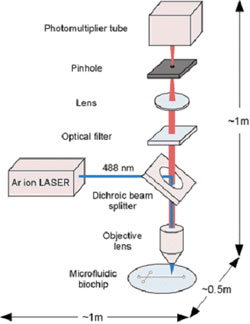 laser-induced fluorescence detection (www.azonano.com).
laser-induced fluorescence detection (www.azonano.com).
MS detection can in principle be applied for all compounds. Determination limits in the ng/mL to high pg/mL level can be obtained, and both qualitative and quantitative (i.e. structure elucidation) data can be obtained.
In a mass spectrometer analytes are determined by measuring the mass-to-charge ratio (m/z). This is done by ionizing the analyte molecules and directing the resulting ions through a series of magnetic and/or electrostatic fields. Four basic types of mass spectrometers are used, in combination with LC, nowadays: magnetic sector, quadrupole, ion-trap and time-of-flight. The magnetic sector instruments use magnetic fields to alter the path of the ionized solute.
Principle of the magnet analyzer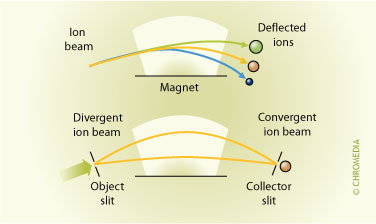 Scanning of molecular weights is performed by changing the strength of the magnetic field.
Scanning of molecular weights is performed by changing the strength of the magnetic field.
The quadrupole instruments are based on the interaction of the ionized solute with an oscillating electric field on four electrodes. By varying the dc and ac potential on the electrodes, only ions of a selected mass will oscillate in a stable path and reach the detector.
In Ion-trap analyzers also an electric field is used, but the ions with stable trajectories are trapped in a cell.
The time-of-flight instrument analyses the time required for ions to reach the detector.
The advantage of the sector instruments is that the molecular weight resolution is relatively high (ca. 0.001 u), the quadrupole instruments are relatively inexpensive, and the time-of-flight instruments have the advantage that there is no upper mass limit of detection. A solute to be analyzed should, therefore, first be vaporized at low pressure, in order to avoid the analyte molecules to react with each other and to facilitate the ionization.
Several ionization modes are in use nowadays, but the two most frequently applied are electron impact (EI) and chemical ionization (CI):
The ionisation principle of the EI technique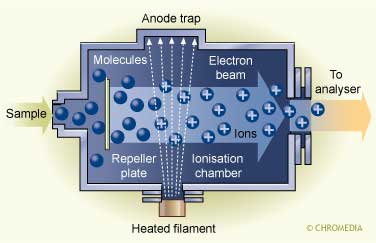
In EI the analytes in the gas phase are bombarded with a beam of electrons resulting in ionization and strong fragmentation of the formed ions. The limitation of EI-MS is that normally only small ions are formed meaning that no information on the molecular weight can be obtained. CI is a milder ionization technique and as a result fragmentation is minimized resulting in molecular weight information. In CI a reagent gas (e.g., methane, ammonia) is ionized by a beam of electrons, and subsequently the analyte is ionized by the transfer of protons from the ionized methane.
The coupling of LC and the MS allies a highly powerful separation technique with a detector possessing unique sensitivity and selectivity (m/z selection; peak patterns) capability. For about a decade, interest has now been focused on the use of LC-MS for the identification and determination of compounds that cannot easily be handled by GC-MS, i.e., compounds with relatively HMW, thermo labile compounds and compounds of a rather polar nature. Unfortunately, the operating conditions peculiar to LC and MS are somewhat incompatible. Consequently, all LC-MS interfaces designed so far impose some restrictions on the normal operation of the LC, the mass spectrometer, or both.
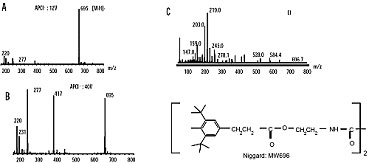 Niggard spectra (polymer additive), obtained by negative APCI (A), negative APCI with in source fragmentation (B), electron impact ionisation (C) (www.waters.com).
Niggard spectra (polymer additive), obtained by negative APCI (A), negative APCI with in source fragmentation (B), electron impact ionisation (C) (www.waters.com).
Gas chromatography can be used for the separation of volatile organic compounds. However, numerous solutes that should be determined in the bio-analytical or environmental field are not suitable for direct GC determination because of there limited stability at the elevated temperatures used during GC separations.
Scheme of a universal GC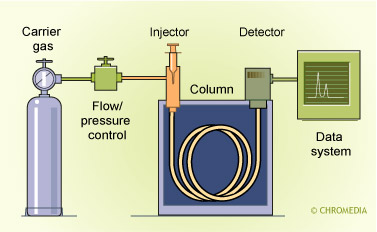
Derivatization with a suitable reagent can significantly improve the temperature stability of an analyte. A Derivatization step should be considered as an integral part of the total ST/ P procedure. During a derivatization reaction normally a polar function of the analyte is converted into a less polar functionality. Derivatization procedures, both for GC and LC, will be discussed extensively in one of the other Chapters of this Topic circle.
SP in GC can be performed nowadays with the traditional techniques like the various modes of extraction, but also multi-dimensional approaches that can be performed either at-line or on-line are widely available. Especially the use of ‘pre-columns’ is gaining in popularity.
GCxGC scheme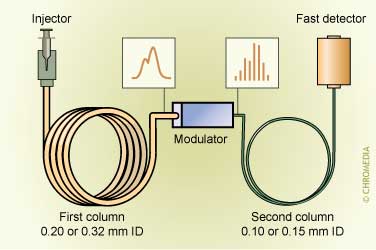
The popularity of GC in environmental analysis is due to fact that GC, in principle, is a more efficient a faster technique – compared with LC – and can be combined easily with a number of selective and sensitive detectors. Moreover, the analytes in environmental analysis are still relatively non-polar compared with the drugs that should be determined in biological samples.
Stationary phases
The key parameter in performing a good GC separation is choosing the optimum stationary phase and column and optimum flow rate of the carrier gas and optimum temperature or temperature program belonging to the chosen set of hardware and the physico-chemical properties of the analyte(s).The fact that the choice of the sorbent in more critical in GC as in LC is due to the fact that in GC no mobile phase is present, the carrier gas mainly act as a transport medium and there are hardly any selective reactions between the analytes and the carrier gas and the carrier gas and the stationary phase.
Rapid temperature programming in gas chromatography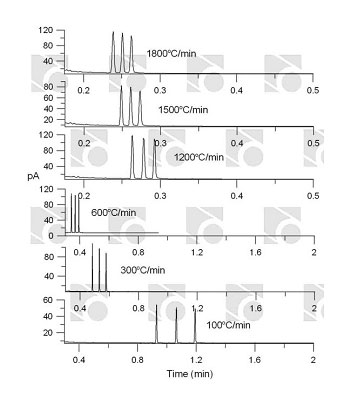 www.rvmscientific.comThe following features are important in choosing the proper separation system:
www.rvmscientific.comThe following features are important in choosing the proper separation system:
- The boiling point range of the analytes and the properties of the resulting vapours phase of these analytes;
- The number of components that must be separated;
- The presence of polar and/or non-polar groups in the analytes and the presence or absence of functional groups;
- The combination of stationary phase and column dimensions as well as the film thickness of the stationary phase with respect to the required selectivity and resolution;
- The flow rate of the carrier gas as a parameter to speed up the total analysis, with a minimum loss in resolution of the most critical pair of compounds to be separated;
- The fact that either a temperature program should be used or that the analysis can be performed at a constant temperature.
Stationary phases in GC can be divided based on their polarity or selectivity with respect to one or more physico-chemical properties of the analyte molecules such as molecular mass or molecular shape, boiling point or the presence of certain functional groups (table herunder).
First of all the stationary phases available for packed columns will be discussed and later on, the nowadays far more popular, sorbents for capillary columns will be discussed.
The separation on the non-polar sorbents like SE-30, OV-1 and Apiezon L, a branched alkane polymer with a melting point of 430C, is primarily based on molecular size and shape. In general the elution order corresponds to the molecular mass of the compounds. In particular for neutral and weakly acidic compounds these phases are suitable.
| Stationary phase | Trade names | Maximum | Applications |
| Polydimethyl siloxane | OV-1, SE-30 | 350 | General-purpose non-polar phase; hydrocarbons; polynuclear aroma-tics; drugs; steroids; PCBs |
| Poly(phenylmethyldimethyl) siloxane (10% phenyl) | OV-3, SE-52 | 350 | Fatty acid methyl esters; alkaloids; drugs; halogenated compounds |
| Poly(phenylmethyl) siloxane (50% phenyl) | OV-17 | 250 | Drugs; steroids; pesticides; glycols |
| Poly(trifluoropropyldimethyl) siloxane | OV-210 | 200 | Chlorinated aromatics; nitroaroma-tics; alkyl-substituted benzenes |
| Polyethylene glycol | Carbowax 20M | 250 | Free acids; alcohols; ethers; es-sential oils; glycols |
| Poly(dicyanoallyldimethyl) siloxane | OV-275 | 240 | Polyunsaturated fatty acids; rosin acids; free acids; alcohols |
Source (D.A. Skoog, D.M. West, F.J. Holler, S.R. Crouch, Fundamentals of Analytical Chemistry, Thomson, Belmont, 2004).
The polyethyleneglycol (PEG) stationary phases are the most frequently used polar sorbents. PEG 400 is a suitable sorbent for the separation of alcohols, ethers, aldehydes and other compounds with low boiling points, while Carbowax 20M can be used for the separation of polar compounds with higher boiling points.
These phases are in particularly useful for the determination of strong basic compounds. Another group of polar stationary phases are the poly(cyanopro-pylphenyldimethyl) siloxanes. These moderately polar sorbents are used for the separation of compounds containing several hydroxyl groups (e.g. steroids).
In summary it can be stated by using only a limited number of sorbents, most separations can be performed in case temperature programming is applied:
| Analytes possessing non-polar groups: | OV-101, Apiezon L |
| Analytes with moderately polar groups: | OV-1, OV-1701 |
| Analytes with polar groups: | Carbowax 20 M. |
As mentioned before different type of columns are used for GC separations:
Cross-section of capillary columns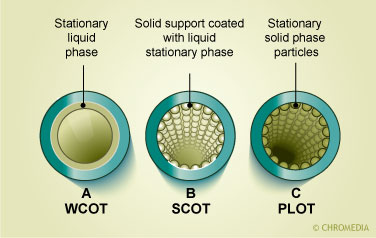
In the picture a packed column with an internal diameter of 2-4 mm and a tubular construction of either glass or stainless steel can be seen. The column is packed with particles which either act as the retarding stationary phase or are coated with a thin film of organic material which acts as the stationary phase. The other pictures depict open tubular columns. These columns have a narrow internal diameter between 0.17-0.53 mm. There are two major types of capillary columns: the porous layer open tubular (PLOT) columns in which the inner surface of the column has an embedded layer that contains the stationary phase and the wall coated open tubular (WCOT) in which a thin film of organic stationary phase is bonded directly onto the inner surface of the column. A ![]() comparison between packed and capillary columns is given in the table.
comparison between packed and capillary columns is given in the table.
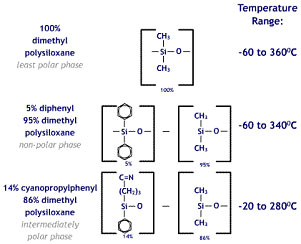 The chemical structure of some of the stationary phases described in this table is pictured here. The non-polar phases are more stable and chromatographically robust than the polar phases which tend to have a lower temperature tolerance and are more susceptible to oxidative damage if air is introduced into the column.
The chemical structure of some of the stationary phases described in this table is pictured here. The non-polar phases are more stable and chromatographically robust than the polar phases which tend to have a lower temperature tolerance and are more susceptible to oxidative damage if air is introduced into the column.
| Stationary phase | Equivalent packed column | Structure of side chains of the poly-siloxane | Polarity | Applications |
| X-1 | OV-101, SE-30 | 100% methyl | Non-polar | Solvents, petroleum pro-ducts, volatile organic com-pounds, environmental con-taminants, amines, drugs |
| X-5 | SE-54 | 5% phenyl, 95% me-thyl | Non-polar | PAHs, perfume components, environmental contaminants, drugs, |
| X-1701, X-10 | OV-1701 | 14% Cyanopropyl, 86% methyl, 50% phenyl | Moderately polar | Pesticides, alcohols, phenols, esters, ketones |
| X-17 | OV-17 | 50% phenyl | Moderately polar | Drugs esters, ketones, plas-ticizers, organochlor com-pounds |
| X-200, X-210 | OV-210 | 50% trifluoropropyl, 50% methyl | Polar | Selective for compounds with free electron pairs, ste-roids, esters, ketones, drugs, alcohols, freons |
| X-WAX | Carbowax 20M | polyethyleneglycol | Strongly polar | Alcohols, methylesters of fatty acids, solvents, fatty acids, amines |
In addition to the excellent separation power of GC another advantage compared with LC is the large number of sensitive and selective detectors that can be used:
| Detector | LOQ | Linear | Temperature | Remarks |
| Thermal conductivity (TCD) | 10-9 | 104 | 450 | Non-destructive, |
| Flame ionization (FID) | 10-12 | 107 | 400 | Destructive, excellent stability |
| Electron capture (ECD) | 10-13 | 103 | 350 | Non-destructive, quickly contaminated, temperature sensitive |
| Nitrogen-phosphor sen-sitive (NPD) | 10-14 | 105 | 400 | See FID |
| Flame photometric (FPD) | 10-12 | 104 |
|
|
| Photo ionization (PID) | 10-12 | 103 |
|
|
There are a number of differences between the detectors used in LC and those in GC. The most important difference is that in LC mainly concentration sensitive detectors are used, while in GC most detection devices are mass sensitive. Some of the GC detectors (i.e. TCD, FID) can be considered to be nearly universal detectors since they respond to a physical change in the carrier gas. The main advantage of the FID (in GC) compared with the refractive-index (RI) detector (in LC) is that the FID is more sensitive and can be used also in combination with temperature programming. Both detectors are considered to be universal.
In addition to the FID, there is a special FID which is selective for nitrogen and phosphor containing compounds, the NPD. In the NPD-mode this detector is about 50 times more sensitive for nitrogen and 500 times for sensitive for phosphor containing compounds.
Another selective detector is the ECD. This detector is frequently used in environmental analysis in case trace analysis of chlorinated pesticides or herbicides should be performed. In addition to the halogenides (F < Cl < Br < I) also a number of organometal compounds (e.g. tetraalkyl lead), NOx and SOx compounds can be selectively determined with this detector. By means of a derivatization procedure with pentafluorophenyl groups, during the SP, a large number of compounds can be determined via GC-ECD.
However, nowadays the most popular detector in combination with a GC separation is the mass spectrometer (GC-MS). This combination allows the separation, identification and quantitation of complex mixtures of compounds. In order for a compound to be determined by GC-MS it should be sufficiently volatile and thermally stable. The procedure is relatively straightforward. The sample is injected into the GC and the analytes are separated on the GC column. The latter part of the column passes through a heated transfer line and ends at the entrance of an ion source, where the analyte molecules, eluting from the column, are converted into analyte ions.
A high voltage separates the charged droplets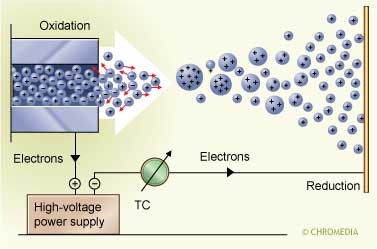 In principle, there are two possibilities for production of the ions. The most frequently used one is electron impact (EI) ionization and in a limited number of cases chemical ionization (CI) is used. For EI a beam of electrons ionize the sample molecules resulting in the loss of one electron. The resulting ion is called the molecular ion and is given by M+. (radical cation). The corresponding peak in the mass spectrum indicates the molecular weight of the compound. Due to the high energy of the electrons, the molecular ions usually fragments into smaller ions with characteristic relative abundances and providing a fingerprint of the molecule. These data can be used to identify analytes and to elucidate the chemical structure of unknown compounds.
In principle, there are two possibilities for production of the ions. The most frequently used one is electron impact (EI) ionization and in a limited number of cases chemical ionization (CI) is used. For EI a beam of electrons ionize the sample molecules resulting in the loss of one electron. The resulting ion is called the molecular ion and is given by M+. (radical cation). The corresponding peak in the mass spectrum indicates the molecular weight of the compound. Due to the high energy of the electrons, the molecular ions usually fragments into smaller ions with characteristic relative abundances and providing a fingerprint of the molecule. These data can be used to identify analytes and to elucidate the chemical structure of unknown compounds.
The next part is the mass analyzer. The mass analyzer is used to separate the positively charged ions according to various mass related properties depending upon the analyzer used.
The magnet bends the ion paths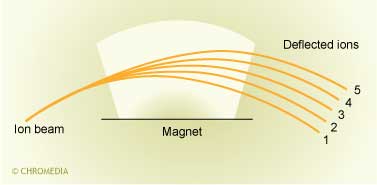
There are several types of analyzers in use. The quadrupoles and the ion-traps are the most frequently applied nowadays.
Only narrow specific m/z values can successfully pass the rods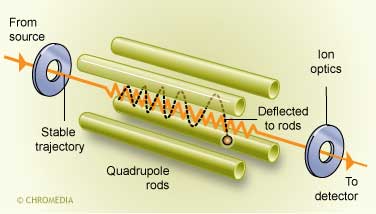
After the ions are separated they enter a detector and after amplification the signal is represented in a mass spectrum in which the intensity of the signal is plotted against the m/z (mass/charge) value.
The power of GC-MS is in the production of mass spectra from each of the analytes detected instead of merely an electronic signal that varies with the amount of analyte. These data can be used to determine the identity as well as the quantity of unknown chromatographic components with an assuredness simple unavailable by other techniques.
The necessity to remove matrix components from the sample to be processed is the normally the most important reason to build in ST/SP steps in an analytical procedure. In most cases this is the only possibility to ensure sufficient selectivity and sensitivity in combination with an electrophoretic or chromatographic separation. In other words, the required reliability can only be obtained when sufficiently clean samples are analyzed.
The objective of the method and subsequently the chosen separation/detection system determine already which ST/SP procedures can or cannot be applied and if manual or automated procedures should be used.
Questions that are important in choosing the proper ST/SP procedures are:
- Should only the analyte itself, or also the degradation products or metabolites be determined? In this case it should be taken into account that degradation products / metabolites are frequently more polar than the parent compound(s).
- Should the free fraction, the matrix-bound fraction or the total amount of the analyte be determined? It may never be assumed that the applied ST/SP procedure will quantitatively disrupt the analyte-matrix bonds.
- Is the analyte enzymatically and/or chemically stable in the matrix? Is it possible to store the samples, should the sample be stabilized or is an immediate analysis necessary?
During method development of an ST/SP procedure there are a number of features that should be taken into account:
- Stability of the analyte;
- Interference by pollutants from reaction- and reagent containers;
- Recovery of the analyte;
- Compatibility of the solvent used in the final ST/SP step and the solvents used in the subsequent separation system;
- Simplicity of the ST/SP procedure;
- Robustness and reliability of the method;
- Possibilities of sample enrichment.
The majority of the available ST/SP procedures can be performed in a manual and automated way. This means that they can be combined off-line, at-line, on-line or in-line with the separation system. In the table an overview is given of frequently applied ST/SP techniques. In addition to the techniques mentioned in this table a filtration or precipitation step is frequently introduced to avoid clogging of the separation ![]() system. The disadvantage of using precipitation techniques is that none of the applied research can be used to precipitate proteins or other macromolecules quantitatively.
system. The disadvantage of using precipitation techniques is that none of the applied research can be used to precipitate proteins or other macromolecules quantitatively.
Sample clean-up in most cases is the time-limiting step in the analytical procedure. The result is that during the past decade a number of methods/procedures have been developed to completely omit a clean-up step in combination with a chromatographic/electrophoretic separation. In a number of cases this can be done by performing the sample clean-up on-line with the separation on a special precolumn.
| Application area | Technique |
| Concentration (enrichment) | Column-switching |
| Removal of high-molecular weight interferences | Column-switching |
| Removal of low-molecular weight interferences | Column-switching |
| Removal of solid particles | Centrifugation |
| Solubilization | Complexation |
One of the approaches is the use of the so-called ‘restricted-access surface’ materials (RAM columns). An example is silica sorbent with relatively small pores. 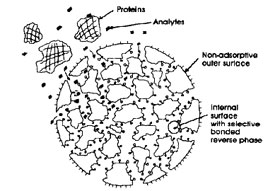 The surface outside of the pores is modified with a hydrophilic phase, while the inside of the pores is coated with a hydrophobic phase.
The surface outside of the pores is modified with a hydrophilic phase, while the inside of the pores is coated with a hydrophobic phase.
Pinkerton ‘Internal-Surface Reversed Phase’ (ISRP) material (www.registech.com).
This type of sorbents has been developed for bio-analytical applications. Proteins and other macromolecules are not able to enter the pores and are excluded from the material. Small molecules, on the contrary, will enter the pores and are retained by means of hydrophobic interactions. The advantage of such an approach is that complex samples can be directly injected into a separation system, which can save quite some time:
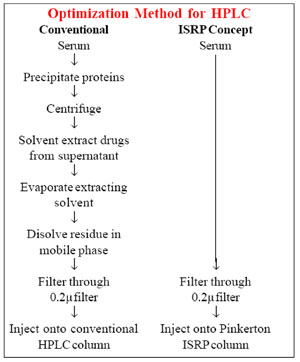 Method development for LC using a restricted-access material (RAM) (www.registech.com).
Method development for LC using a restricted-access material (RAM) (www.registech.com).
In case relatively small samples series (< 25) should be analyzed once, or only a few times a year, a combination of a precipitation or filtration procedure with an off-line LLE or SPE seems to be the best approach. In case larger numbers of samples should be analyzed sample processors and flow-injection devices – semi-flexible automated systems which can perform a limited number of manipulations, column-switching techniques – ![]() automated systems which can perform only one particular application, and robotic units – flexible automated systems which can perform nearly all of the known ST/SP manipulations.
automated systems which can perform only one particular application, and robotic units – flexible automated systems which can perform nearly all of the known ST/SP manipulations.
Sample processor. A sample processor can perform a limited number of  manipulations (e.g., pipetting, diluting, mixing, SPE) in an automated way, is relatively cheap,
manipulations (e.g., pipetting, diluting, mixing, SPE) in an automated way, is relatively cheap, ![]() flexible in use and method development is relatively fast:
flexible in use and method development is relatively fast:
Sample processor (www.hnp-mikrosysteme.de).
Column-switching techniques: By using one or more switching valves several columns can used in series or in parallel. In this way numerous options are available to improve the selectivity and/or the sensitivity of a procedure. Important features of such an approach are: high reproducibility but limited flexibility. For every single application another column-switching system must be used and method development is rather time-consuming:
Switch positions of valco valve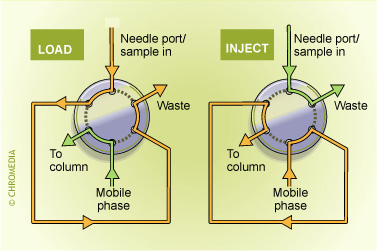
Flow-injection devices: Flow-injection analysis (FIA) is a well known on-line approach allowing simple manipulations (e.g., LLE, dialysis, concentration) in a reproducible way. FIA is a flexible approach and is relatively cheap. Method development normally is relatively fast.
Robotic systems: A robot can, in principle, perform all manipulations  needed in a chemical analytical process. The advantages of a robot system, over human manipulations, are the fact that extreme conditions and extended working hours are not causing any problems. The most important features of robotic systems are: high costs, reproducibility and flexibility, and time-consuming method development procedures.
needed in a chemical analytical process. The advantages of a robot system, over human manipulations, are the fact that extreme conditions and extended working hours are not causing any problems. The most important features of robotic systems are: high costs, reproducibility and flexibility, and time-consuming method development procedures.
Robotic systems (www.beckman.com).
A general conclusion is that automation in order to limit the number of manual sample manipulations steps will significantly increase the complexity and the costs of the total analytical procedure.
The three problems given in this section are representative for the problems that can be solved after studying the material provided in this module on “Structure Related Sample Treatment”.
| Problem 1: Bio-analytical problem |
| A 45 year old man is probably injected with Unasyn (ampicillin 1,000 mg + sulbactam 500 mg) directly in the vein. After about 45 min the man died because of presumed anaphylaxis. Meanwhile he was taking nimesulide, piroxicam, tiocolchicoside, claritromicina and maybe lidocain or other drugs in the vein. |
| Problem 2: Bio-analytical problem |
| The active compounds in a certain type of mushrooms (that are used in the drugs scene) are psilocine and psilocybine. Psylocibine is more active compared with psilocine. However, in the body psilocybine is converted to psilocine. Psilocine is structurally related to serotonine and is blocking the serotonine receptors on the dendrites. The serotonine that, after stimulation of a neuron, is entering the synaptic gap, is taken up again by the axon endings. The result of a low serotonine level is an increase of the dopamine level. After some time the psilocine is enzymatically degraded and the receptors are able to take up serotonine again. At the moment it is not completely clear how a change in the serotonine- and dopamine regutation results in hallucination and other sensations. It is clear, however, from brain scans that the area of data handling is shifting from the left (rational-analytical) to the right (visual-spacial) brain part under the influence of hallucinogenic compounds.
|
| Problem 2.3: Bio-analytical problem |
| A serum sample is arriving in the laboratory with the following information: The patient is treated with a relatively new analgesic agent (10.5). The therapeutic range of this compound is relatively small. The necessary serum levels are between 300 – 500 ng/mL. Toxic effects are already observed at concentrations of 650 ng/mL.
|






