Abstract This chapter describes the properties of hybrid stationary phases, with special emphasis on their use in method development. A key feature of these phases is their stability at alkaline pH, which expands their usefulness in method development. It also describes how analyte features influence retention as a function of pH, column, and mobile phase.
LevelAdvanced
Hybrid Stationary Phases for RPLC and the Use of these Packings in Method Development.
Classically, reversed-phase packings have been created by modifying the surface of silica with hydrophobic ligands. While much improvement has been made with this technology over the years [![]() 1], the use of conventional bonded phases based on silica was limited at alkaline pH [ 2 , 3, 4]. On the other hand, polymer-based packings such as styrene-divinylbenzene-based packings, which are completely stable at alkaline pH, had a range of weaknesses, including difficulties with the mechanical stability and an inferior mass-transfer. Particles based on other inorganic oxides such as alumina or zirconia [5, 6] have a good stability in the alkaline pH range, but the surface chemistry of these oxides is more complicated than that of silica. Since for these oxides there is no straightforward bonding to the surface available as for silica, such phases require a polymer coating with consequently slow mass transfer, or other special coating techniques which result in surface properties significantly different from a standard C18. At the same time, it was obvious that an exploration of the alkaline pH-range would expand significantly the range of parameters that the method development chemist could work with [4
1], the use of conventional bonded phases based on silica was limited at alkaline pH [ 2 , 3, 4]. On the other hand, polymer-based packings such as styrene-divinylbenzene-based packings, which are completely stable at alkaline pH, had a range of weaknesses, including difficulties with the mechanical stability and an inferior mass-transfer. Particles based on other inorganic oxides such as alumina or zirconia [5, 6] have a good stability in the alkaline pH range, but the surface chemistry of these oxides is more complicated than that of silica. Since for these oxides there is no straightforward bonding to the surface available as for silica, such phases require a polymer coating with consequently slow mass transfer, or other special coating techniques which result in surface properties significantly different from a standard C18. At the same time, it was obvious that an exploration of the alkaline pH-range would expand significantly the range of parameters that the method development chemist could work with [4![]() ].
].
This led a group of scientists a Waters Corporation to explore technologies that could result in packings with improved pH stability retention properties similar to classical silica-based packings [7![]() ]. Inorganic-organic hybrid materials had proven to be successful in many technology areas, and there was promise that such materials could meet the goal of creating alkaline-stable packings with an overall strong similarity to conventional silica-based reversed-phase packings. A few years later, after some solid research, the first of these packings was introduced to the chromatographers.
]. Inorganic-organic hybrid materials had proven to be successful in many technology areas, and there was promise that such materials could meet the goal of creating alkaline-stable packings with an overall strong similarity to conventional silica-based reversed-phase packings. A few years later, after some solid research, the first of these packings was introduced to the chromatographers.
Let us define what inorganic-organic hybrids are in the context of this paper. In general, there is a vast number of possibilities that can be used to combine organic and inorganic components. We would like to define hybrids in this context as the combination of inorganic and organic components at the molecular level [7]. In HPLC, the classical substrate for bonding technology is silica. Modern, high-purity silicas are prepared from tetraethoxy silane or similar hydrolysable silanes of high purity. For the preparation of hybrid packings for HPLC, the tetraethoxy silane is co-reacted with suitable organosilanes, for example methyltriethoxy silane. This co-reaction at the molecular level means that the final product is not simply a blend, but a new chemical entity with novel – and as it turns out – very useful chemical properties.
A range of different organosilanes with chemically stable silicon-carbon bonds can be envisioned as partners in this reaction [7]. Some of these silanes provide hydrophobicity to the substrate, others incorporate new functional groups that can be used as the basis for new surface-bonding techniques. At this moment, only a few options for the design of such phases have been realized commercially. We will describe these phases here in detail, together with their use in chromatography, especially in methods development.
The synthesis of hybrid packings is similar to the preparation of a modern high-purity silica. The preparation method proceeds in several steps. First, the organo-functional silane is coreacted with tetraethoxysilane to form an oligosiloxane precursor with a controlled molecular weight distribution. This oligosiloxane is then used in the actual particle preparation process to create HPLC particles with a controlled pore volume and pore size distribution. Particle-size distributions useful for packing HPLC columns are then generated using suitable classification techniques such as air classification.
Two types of hybrid packings are commercially available at the time of this writing, a methyl hybrid packing with methyl groups incorporated into the matrix of the packing (XTerra®), and a packing with an ethyl bridge in the matrix (XBridge®). The details of the synthesis of the latter are shown in figure 1.
1. Synthesis scheme for ethyl-bridged hybrid particles.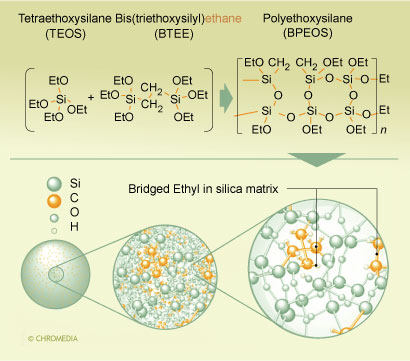 U. Neue, Waters
U. Neue, Waters
These hybrid packings still contain silanols on the surface that can then be derivatized in a second step with the same technology and the same ligands as is suitable for silica. One can derivatize with mono-, di-, or tri-functional silanes with the appropriate functions to obtain C18, C8, phenyl or other surfaces, such as bonded phases with embedded polar groups. For example, for the ethyl-bridged hybrid packing, the bonded phases and ligands available are shown in figure 2. The surface coverage of the different ligands is uniformly around 3.2 µmoles/m2. We will show later how the properties of these different bonded phases can be used in method development.
2. Commercial bonded phases for the ethyl-bridged hybrid packing.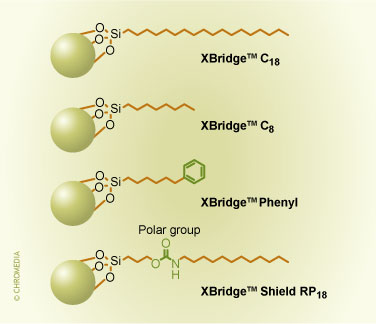
There are two important features of these hybrid packings that play a critical role for the chromatographer. One is the significant improvement in the stability of such packings in the alkaline pH range [8![]() , 9], the other is the shift in silanol activity compared to classical silica-based packings. The ethyl-bridged hybrid packing has been tested in applications using a 20 mM phosphate buffer at pH 12. It was demonstrated that the column survived over 700 20-minute analyses at 30°C in such a buffer. In modern HPLC with MS detection, a more commonly used buffer is the volatile ammonium bicarbonate buffer at pH 10, and under these conditions the stability of the packing is as good as the stability of silica-based packings in the common pH range used for silicas. This opens the opportunity to indeed explore the impact of the alkaline pH on separation and detection using packings with essentially the same surface properties as classical silica-based packings [9]. This indeed has been done in many applications. We will discuss a possible method development scheme in the next section.
, 9], the other is the shift in silanol activity compared to classical silica-based packings. The ethyl-bridged hybrid packing has been tested in applications using a 20 mM phosphate buffer at pH 12. It was demonstrated that the column survived over 700 20-minute analyses at 30°C in such a buffer. In modern HPLC with MS detection, a more commonly used buffer is the volatile ammonium bicarbonate buffer at pH 10, and under these conditions the stability of the packing is as good as the stability of silica-based packings in the common pH range used for silicas. This opens the opportunity to indeed explore the impact of the alkaline pH on separation and detection using packings with essentially the same surface properties as classical silica-based packings [9]. This indeed has been done in many applications. We will discuss a possible method development scheme in the next section.
Another interesting consequence of the design of hybrid packings is the reduction in silanol activity, which is partially due to the lower population of surface silanols, and partially due to a shift of the pKa of the surface silanols. The former is simply due to the presence of the organic component in the matrix, which reduces the amount of silanols that could be present on the surface. The latter has first been pointed out in reference 10, and was confirmed later in reference 11. For the methyl-hybrid packing XTerra®, there appears to be a rather large population of methyl groups on the surface, since surface bonding is limited to only about 2.5 µmol/m2. At the same time, the silanol activity of the packing is very low. The best interpretation of both these facts is a very low silanol population on the surface before bonding. In addition, the increase in the pK of the surface silanols from values of around 7 for high purity silicas to values around 9 for the methyl-hybrid packing speaks for the influence of the hydrophobic environment on the pKa of these surface silanols. The surface coverage for the ethyl-bridge hybrid packing is higher, but still not as high as would be possible on a silica surface. The reduction in silanol activity is a benefit with respect to the peak shape of basic analytes, which is a problem with silica-based packings based on the older lower-purity silicas.
Overall, hybrid packings have a drastically improved stability at high pH compared to silica-based packings, and a lower silanol activity. Both properties are attractive features for method development, which will be discussed in the next section.
Method development is never an easy task, and chemists involved in it welcome every new tool that can help them solving problems. The principle that we are proposing uses several different ideas and combines them into a single approach. The classical method development scheme is based on the use of different solvents to achieve different selectivity [12, ![]() 13]. If packings are available that deliver a good peak shape for all analytes, as it is possible with modern high-quality stationary phases, then one can add pH as a powerful and independent dimension in method development [14, 15
13]. If packings are available that deliver a good peak shape for all analytes, as it is possible with modern high-quality stationary phases, then one can add pH as a powerful and independent dimension in method development [14, 15![]() , 16]. In addition, different stationary phases give differences in selectivity [16, 17]. If a good process can be developed that explores these three major influences on the selectivity of a separation in a rapid and efficient way, it would be a very useful tool for a general approach to explore the selectivity options available. This exactly is possible with the use of hybrid packings.
, 16]. In addition, different stationary phases give differences in selectivity [16, 17]. If a good process can be developed that explores these three major influences on the selectivity of a separation in a rapid and efficient way, it would be a very useful tool for a general approach to explore the selectivity options available. This exactly is possible with the use of hybrid packings.
We have developed a method screening process that explores simultaneously solvents, pH and the packing materials chosen to give the largest selectivity differences possible within reversed-phase chromatography (see below). The principle is shown in figure 3. One uses three column types that are known to give a significant difference in selectivity, i.e. a C18 column, a column with an embedded polar group and a phenyl column [![]() 16, 17, 18]. We have used two sets of columns that fulfill this selectivity pattern and are also stable at alkaline pH: XTerra MS C18, XTerra RP18 (the packing with the embedded polar group) and XTerra Phenyl, or XBridge C18, the EPG packing XBridge RP18 and XBridge Phenyl [18]. As the two solvent choices, we have used acetonitrile and methanol [16, 19]. The choices of the buffers for the control of the pH are determined by the choice of the detector. Since today most methods need to be MS compatible, the primary choices are formic acid or an ammonium formate buffer around pH 3.75, and an ammonium bicarbonate buffer at pH 10 [16, 17]. Ammonium bicarbonate has the advantage of combining the buffer capacity of the ammonium ion with that of the carbonate ion, but it can also be replaced with an ammonium formate buffer at pH 9.25, or with ammonia, for example in preparative applications. Of course, other options are conceivable, and other detection schemes may dictate a different buffer choice.
16, 17, 18]. We have used two sets of columns that fulfill this selectivity pattern and are also stable at alkaline pH: XTerra MS C18, XTerra RP18 (the packing with the embedded polar group) and XTerra Phenyl, or XBridge C18, the EPG packing XBridge RP18 and XBridge Phenyl [18]. As the two solvent choices, we have used acetonitrile and methanol [16, 19]. The choices of the buffers for the control of the pH are determined by the choice of the detector. Since today most methods need to be MS compatible, the primary choices are formic acid or an ammonium formate buffer around pH 3.75, and an ammonium bicarbonate buffer at pH 10 [16, 17]. Ammonium bicarbonate has the advantage of combining the buffer capacity of the ammonium ion with that of the carbonate ion, but it can also be replaced with an ammonium formate buffer at pH 9.25, or with ammonia, for example in preparative applications. Of course, other options are conceivable, and other detection schemes may dictate a different buffer choice.
3.Rapid selectivity screening: first step in method development.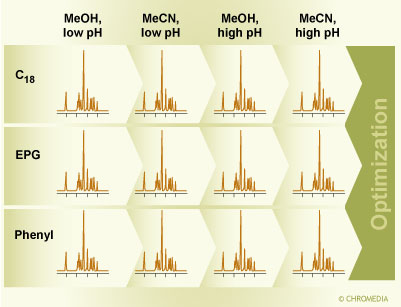 Figure 3: Rapid selectivity screening as the first step in method development. Three selected column types are used, a C18 column, a phenyl column, and a column with embedded polar group using methanol and acetonitrile at high and low pH.
Figure 3: Rapid selectivity screening as the first step in method development. Three selected column types are used, a C18 column, a phenyl column, and a column with embedded polar group using methanol and acetonitrile at high and low pH.
The combination of columns, organic solvents and buffers requires 12 experiments for a complete screening of all options [16]. Using short, 5 cm columns, rapid 20 minute gradients, and MS detection for the identification of the analytes, such a method screening can be carried out in a short amount of time. One then ends up with 12 chromatograms that can be examined for selectivity differences and resolution. This process can not only be carried out, when standards are available. It is also possible to do this with an unknown sample, such as a forced degradation of a pharmaceutical, especially if MS identification of the peaks is available. The technique can also be used in cases, where an optimal separation of the analytes of interest from matrix interferences is desired [16]. Such situations arise with many sample types, from environmental waters to food to blood samples.
A detailed procedure for this method exploration [from reference 16] is given in this paragraph, and we will discuss the reasons for the choices in the next section. Use 4.6 mm x 50 mm columns packed with 3.5 or 5 µm packings. The selected packings are a classical C18 or C8 column, a column with an embedded polar group, and a phenyl packing. The organic modifiers are methanol and acetonitrile, and the buffers are ammonium formate at pH 3.75 and ammonium bicarbonate at pH 10 at a concentration of 10 mM. Use a flow rate of 2 mL/min and run gradients from 0 to 80% organic solvent over a gradient run time of at least 15 minutes. You will obtain 4 chromatograms for every column. If you need to identify the peaks with standards, you will need additional chromatograms for that purpose. As the end result of this study, you will have 12 chromatograms with identified peaks that you can compare against each other for best results. A simple visual side-by-side inspection of the 12 chromatograms is best for the selection of the next step. If you have less than 10 compounds to separate, and if these compounds respond to the pH changes, the chances are high that you will have a suitable separation in your hands at the end of this blind activity. If the separation is more complex or requires additional optimization, the screening exercise will have given you the best starting point(s) for further action. Note that this combination of parameters was designed to maximize the selectivity differences that one can encounter in reversed-phase chromatography. Consequently, it is less likely that the same good results can be achieved just with different C18 columns from different manufacturers under acidic pH conditions, as is advocated in other method development schemes.
As a next step, it is often useful to flatten or sharpen some sections of the gradient or to obtain chromatograms with mixtures of acetonitrile and methanol as the organic modifiers. A fine tuning of the pH might be useful, or the selection of a different temperature. Unless the original chromatogram from the screening is very promising, the use of temperature as the next variable is the simplest choice for finetuning or further optimization of the method.
The choices of mobile phases, pH and columns are not arbitrary, but have a solid foundation. In a large study with over 70 different analytes, the influences of solvent pH and column choice were examined ![]() [18, 19]. I will summarize some of the findings here.
[18, 19]. I will summarize some of the findings here.
If we have a mixture of acids, bases and zwitterionic analytes, pH has the strongest impact on the selectivity of a separation [Figure 4]. The retention of the same analyte in the ionic form and in the non-ionic form changes at least 10-fold, often 30-fold in the case of small molecules such as pharmaceuticals. Acids are more retained at acidic pH, and bases are more retained at basic pH. However, even if one needs to separate only analytes of the same type, i.e. only acids or only bases, there is a significant difference in the selectivity of the separation between both forms of the same molecule. The reason for these differences in selectivity can be explained as follows. It is possible, that the ionic site of the analyte binds water strongly, and that this hydrated portion of the molecule is hidden from interaction with the hydrophobic ligand on the surface of the packing. If at the opposite pH the charge is lost, the strongly bound water layer is lost as well, and the surroundings of the formerly strongly hydrated ionic sites can now bind to the hydrophobic ligand. It is also possible that the ionization of these functional groups impede the penetration of the analyte into the bonded-phase layer. Also, changes in the structure of the analyte are possible as a function of the charges on the molecule. These effects alone can lead to significant selectivity differences between the different pH values, but one can also fine-tune the pH to manipulate the retention of partially ionized analytes.
4: Comparison of the gradient retention times 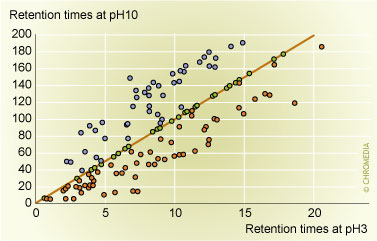 Figure 4: Comparison of the gradient retention times of analytes at pH 10 and pH 3. Neutral analytes, shown in purple, have the same retention at both pH values. Basic analytes, shown in blue, exhibit more retention at alkaline pH, while acidic analytes, in red have more retention at acidic pH.
Figure 4: Comparison of the gradient retention times of analytes at pH 10 and pH 3. Neutral analytes, shown in purple, have the same retention at both pH values. Basic analytes, shown in blue, exhibit more retention at alkaline pH, while acidic analytes, in red have more retention at acidic pH.
The differences in retention for solvents and columns are not as drastic as those associated with pH. On the other hand, pH changes have no influence whatsoever on the retention of neutral analytes, but solvents and columns do. In all cases, but even more so for the latter case, the choice of solvent and column type influences retention and selectivity. We examined different columns with the different analytes mentioned above in methanol and acetonitrile and at three different pH values and plotted the resulting 420 retention data for each different column against each other. These plots gave different levels of data scatter, which then can be used to judge the selectivity difference between different columns. When comparing a C8 to a C18 column, the scatter was rather small: the selectivity differences observed were not very large. The scatter was significantly larger for a comparison of a C18 column to a packing with an embedded polar group, or to a packing with a phenyl ligand. The amount of scatter can be quantitated [17, 18, 19], and this resulted in the choice of a C18 column, a phenyl column and a column with an embedded polar group as the column choices with the largest selectivity differences in our method development scheme.
These overall selectivity differences between the different packings can be examined further. It has been known since a long time that π-π interactions are the important feature of phenyl packings. Indeed, it can be demonstrated that analytes with long chains show more retention on a C18 packing, while analytes that exhibit π-π interactions with the phenyl ligand exhibit more retention on a phenyl packing [18]. It is interesting that positively charged amines also have a preferred interaction with the electron-rich π-groups. With respect to EPG packings, we have specifically studied the packings with an incorporated carbamate groups. These packings give more retention for analytes with non-ionized phenol and carboxylic acid groups, and sulfonamides. This is due to hydrogen bonding between these functional groups and the carbamate group. Also, positively charged amines are much less retained on EPG packings. This is believed to be due to the effective shielding of residual silanols by a layer of water tightly bound by the embedded polar group.
The influence of the choice of the organic modifier on the selectivity of a separation is about as large as the effects of well-chosen columns, but it is more difficult to assess. The reason is that the organic solvent is both present in the stationary phase and in the mobile phase, and that selectivity differences between solvents arise from differences in the solvation of the analyte between both phases. On the other hand, the difference in the elution strength of the different modifiers has been known since a long time. Methanol is a weaker eluent than acetonitrile, which in turn is weaker than THF. However, a few more specific rules become clear, when a large group of analytes is used [19]. For example, ionized analytes are relatively more retained with methanol as the organic modifier, compared to acetonitrile. Sulfonamides have a comparatively larger retention in THF compared to methanol. Amines appear to be comparatively better retained in methanol than in THF. The plot of the solvent strength that gives equal retention between acetonitrile and methanol is curved for classical C18 and C8 phases, but linear for packings with an embedded polar group. On the other hand, there is a linear solvent-strength relationship between THF and methanol on the classical phases, and a curved relationship for phases with an embedded polar group. Overall, it is noteworthy that THF gives the largest selectivity differences, but we did not include it in our method development scheme, since many chromatographers do not use THF due to its unpleasant odor and the potential of the formation of peroxides. Sufficient selectivity differences are observed between methanol and acetonitrile.
1. U. D. Neue, "Silica Gel and its Derivatization for Liquid Chromatography", in "Encyclopedia of Analytical Chemistry", R. A. Meyers, Ed., John Wiley & Sons, Ltd., Chichester (2000), 11450-11472
2. J. J. Kirkland, M. A. van Straten, H. A. Claessens, ”High pH mobile phase effects on silica-based reversed-phase high-performance liquid chromatographic columns”, J. Chromatogr. A 691 (1995), 3-19
3. H. A. Claessens, M. A. van Straten, J. J. Kirkland, ”Effect of buffers on silica-based column stability in reversed-phase high-performance liquid chromatography”, J. Chromatogr. A 728 (1996), 259-270
4. J. J. Kirkland, J. W. Henderson, J. J. DeStefano, M. A. van Straten, H. A. Claessens, ”Stability of silica-based, endcapped columns with pH 7 and 11 mobile phases for reversed-phase high-performance liquid chromatography”, J. Chromatogr. A 762 (1997), 97-112
5. J. Nawrocki, C. Dunlap, A. McCormick, P. W. Carr, “Part I: Chromatography using ultr-stable metal oxide-based stationary phases for HPLC”, J. Chromatogr A 1028 (2004), 1-30
6. J. Nawrocki, C. Dunlap, J. Li, J. Zhao, C. V. McNeff, A. McCormick, P. W. Carr, “Part II: Chromatography using ultr-stable metal oxide-based stationary phases for HPLC”, J. Chromatogr A 1028 (2004), 31-62
7. J. E. O’Gara, K. D. Wyndham, “Porous Hybrid Organic-Inorganic Particles in Reversed-Phase Liquid Chromatography”, J. Liq. Chromatogr. Rel. Technol. 26 (2006), 1025-1045
8. K. D. Wyndham, J. E. O’Gara, T. H. Walter, K. H. Glose, N. L. Lawrence, B. A. Alden, G. S. Izzo, C. L. Hudalla, P. C. Iraneta, “Characterization and Evaluation of C18 HPLC Stationary Phases Based on Ethyl-Bridged Hybrid Organic/Inorganic Particles”, Anal. Chem. 75 (2003), 6781-6788
9. Y.-F. Cheng, T. H. Walter, Z. Lu, P. Iraneta, B. A. Alden, C. Gendreau, U. D. Neue, J. M. Grassi, J. L. Carmody, J. E. O’Gara, R. P. Fisk, “Hybrid organic-inorganic Particle Technology: Breaking through Traditional Barriers of HPLC Separations”, LC/GC 18 (2000), 1162-1172
10. U. D. Neue, C. H. Phoebe, K. Tran, Y.-F. Cheng, Z. Lu, “Dependence of Reversed-Phase Retention of Ionizable Analytes on pH, Concentration of Organic Solvent and Silanol Activity”, J. Chromatogr. A 925 (2001), 49-67Retention of Ionizable Analytes on pH, Concentration of Organic Solvent and Silanol Activity”, J. Chromatogr. A 925 (2001), 49-67
11. A. Méndez, E. Bosch, M. Rosés, U. D. Neue, “Comparison of the acidity of residual silanol groups in several liquid chromatography columns”, J. Chromatogr. A 986 (2003), 33-44
12. J. L. Glajch, J. J. Kirkland and K. M. Squire, J. M. Minor, “Optimization of solvent strength and selectivity for reversed-phase liquid chromatography using an interactive mixture-design statistical technique”, J. Chromatography 199 (1980). 57-79
13. J. L. Glajch, J. J. Kirkland, L. R. Snyder, “Practical optimization of solvent selectivity in liquid-solid chromatography using a mixture-design statistical technique”, J. Chromatography 238 (1982), 269-280
14. U. D. Neue, "HPLC-Columns - Theory, Technology, and Practice", Wiley-VCH, 1997
15. Y.-F. Cheng, M. Z. El Fallah, U. D. Neue, D. J. Phillips, “Simple and Fast Methods Development for HPLC and Sample Preparation”, Waters Column VI (5) (1997), 10-15
16. U. D. Neue, E. S. Grumbach, J. R. Mazzeo, K. Tran, D. M. Wagrowski-Diehl, “Method development in reversed-phase chromatography”, Handbook of Analytical Separations Vol. 4, pp. 185-214, I. D. Wilson ed., Elsevier Science, 2003
17. U. D. Neue, A. Méndez, K. Tran, D. M. Diehl, “pH and Selectivity in Reversed-Phase Chromatography”, in “HPLC Made to Measure: A Practical Handbook for Optimization”, Stavros Kromidas, Ed., pp. 59-75, Wiley-VCH, Weinheim, 2006
18. U. D. Neue, J. E. O’Gara, A. Méndez, “Selectivity in Reversed-Phase Separations: Influence of the Stationary Phase”, J. Chromatogr. A 1127 (2006), 161-174
19. U. D. Neue, A. Méndez, “Selectivity in Reversed-Phase Separations I. General Influence of Solvent Type and Mobile Phase pH”, J. Sep. Sci., accepted for publication





